Highly Potent, Selective Acylguanidine-Type Histamine H2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
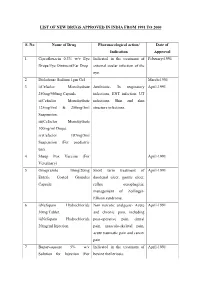
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

SYNTHESIS and STRUCTURE ACTIVITY STUDIES of NOVEL H3-RECEPTOR HISTAMINE ANTAGONISTS. a Thesis Presented in Partial Fulfilment Of
1 SYNTHESIS AND STRUCTURE ACTIVITY STUDIES OF NOVEL H3-RECEPTOR HISTAMINE ANTAGONISTS. A thesis presented in partial fulfilment of the requirements for the Doctor of Philosophy Degree of the University of London SEYED KIUMARS HOSSEINI Department of Chemistry University College London September 1991 Attention is drawn to the fact that copyright of this thesis rests with its author. This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that the copyright rests with its author and that no quotation from the thesis and no information derived from it may be published without prior consent of the author. Signature: Date..^*^^~ ProQuest Number: 10609847 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 10609847 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 2 Acknowledgements I would like to express my gratitude for the guidance and generous support of Prof. C. R. Ganellin and for the financial support of Bioproject in association with Centre Paul Broca de rinserm, Paris. Thanks to Prof. -

Human Histamine H2 Receptor, Frozen Cells Product No.: ES-391-AF
TECHNICAL AequoZen® DATA SHEET Research use only. Not for use in diagnostic procedures. You are authorized to utilize these frozen cell preparations one time only. Any attempt to transfer, re-use, or propagate these cells is expressly unauthorized and a violation of the product terms and conditions of sale. Human Histamine H2 Receptor, Frozen Cells Product No.: ES-391-AF Lot No.: 2562845 Material Provided Cells: 1 x 1 mL frozen aliquot Format: ~10 x 106 cells/mL in Ham’s F12, 10% FBS with 10 % DMSO Product Information Cellular Background: CHO-K1 Parental Frozen Cells (control): A19 (replaced with Cat # ES-000-A2F) Frozen Cells Info: Frozen recombinant, CHO-K1 cells expressing mitochondrially- targeted Aequorin, Gα16 and the human Histamine H2 receptor. DNA Sequence: Identical to coding sequence of GenBank NM_022304.2. Corresponding Protein Sequence: Identical to GenBank NP_071640.1. Storage Conditions: Store in liquid nitrogen (vapor phase) immediately upon receipt, or maximum 15 days at -80°C. AequoZen® is designed for single use only. Do not refreeze. Quality Control ® EC50 for a reference agonist is determined using an AequoScreen assay (Figure 1). Mycoplasma test is performed using MycoAlert® Mycoplasma detection kit. We certify that these results meet our quality release criteria. Amthamine dihydrobromide (EC50): 6.9 nM Mycoplasma: This cell line tested negative for Mycoplasma. TDS-ES-391-AF-04 Page 1 of 5 Recommended Thawing Conditions and Handling of Frozen Cells Carefully follow instructions below to obtain the expected results. Most Frozen cells are intended to be assayed immediately upon thawing. Exceptionally, where specified, some frozen cell products require an overnight incubation in Cell Medium to enable them to perform optimally. -

Chapter 2 Molecular Aspects of Histamine Receptors
VU Research Portal Shedding Light on the Histamine H3 Receptor Mocking, T.A.M. 2020 document version Publisher's PDF, also known as Version of record Link to publication in VU Research Portal citation for published version (APA) Mocking, T. A. M. (2020). Shedding Light on the Histamine H3 Receptor: Photopharmacology and bioluminescent assays to study GPCRs. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 06. Oct. 2021 Chapter 2 Molecular aspects of histamine receptors Histamine mediates a multitude of physiological effects in the human body by activating four histamine receptor subtypes. Histamine receptors have proven to be promising drug targets in the treatment of a variety of diseases, including hay fever, gastric ulcers, inflammatory and neuropathological diseases. In this chapter the molecular aspects of histamine receptors are described, including expression profile, intracellular signaling, and how histamine receptor activity can be attenuated by ligands targeting the histamine receptor binding sites. -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Pharmacology and Toxicology of Metiamide, a Histamine H
9 November 1974 S.-A. MEDIESE TYDSKRIF 2253 Pharmacology and Toxicology of Metiamide, a - Histamine H2 Receptor Antagonist R. W. BRIMBLECOMBE, W. A. M. DUNCAN, M. E. PARSONS SUMMARY x 10-'M on atrial muscle and 7,5 x lO-rM on uterine muscle. Even. at lO-'M, metiamide did not inhibit the A brief review of the pharmacology and toxicology of effects of isoprenaline on either of these tissues neither did it inhibit the effects of histamine on isolated Quinea- metiamide, a histamine H2-receptor antagonist, is given, ~ and evidence is presented to support the view that it pig ileum (mediated through H,-receptors). inhibits gastric acid secretion by virtue of its Hrreceptor Metiamide is also an inhibitor of gastric acid secretion. antagonist activity. Its effectiveness was estimated in two preparations: the Studies are also reported which show that metiamide lumen-perfused stomach of the anaesthetised rat' and given either intravenously or intraduodenally inhibits his the conscious Heidenhain pouch dog, prepared 1 - 3 years tamine- or pentagastrin-stimulated acid secretion in human before experimentation. Metiamide was given by rapid subjects. llltravenous injection during a maximal plateau of acid secretion stimulated by either histamine or pentagastrin, and the dose required to reduce this level of secretion by S. Afr. Med. J., 48, 2253 (1974). 50% (ED",) was estimated. The results are shown in Table I and indicate that the ED", values are very similar Conventional antihistaminic drugs, such as mepyramine. even in high concentrations, fail to inhibit histamine against those of both histamine and pentagastrin. Doses of stimulated gastric acid secretion. -

Histamine Receptors
Tocris Scientific Review Series Tocri-lu-2945 Histamine Receptors Iwan de Esch and Rob Leurs Introduction Leiden/Amsterdam Center for Drug Research (LACDR), Division Histamine is one of the aminergic neurotransmitters and plays of Medicinal Chemistry, Faculty of Sciences, Vrije Universiteit an important role in the regulation of several (patho)physiological Amsterdam, De Boelelaan 1083, 1081 HV, Amsterdam, The processes. In the mammalian brain histamine is synthesised in Netherlands restricted populations of neurons that are located in the tuberomammillary nucleus of the posterior hypothalamus.1 Dr. Iwan de Esch is an assistant professor and Prof. Rob Leurs is These neurons project diffusely to most cerebral areas and have full professor and head of the Division of Medicinal Chemistry of been implicated in several brain functions (e.g. sleep/ the Leiden/Amsterdam Center of Drug Research (LACDR), VU wakefulness, hormonal secretion, cardiovascular control, University Amsterdam, The Netherlands. Since the seventies, thermoregulation, food intake, and memory formation).2 In histamine receptor research has been one of the traditional peripheral tissues, histamine is stored in mast cells, eosinophils, themes of the division. Molecular understanding of ligand- basophils, enterochromaffin cells and probably also in some receptor interaction is obtained by combining pharmacology specific neurons. Mast cell histamine plays an important role in (signal transduction, proliferation), molecular biology, receptor the pathogenesis of various allergic conditions. After mast cell modelling and the synthesis and identification of new ligands. degranulation, release of histamine leads to various well-known symptoms of allergic conditions in the skin and the airway system. In 1937, Bovet and Staub discovered compounds that antagonise the effect of histamine on these allergic reactions.3 Ever since, there has been intense research devoted towards finding novel ligands with (anti-) histaminergic activity. -

Tall Man Lettering List REPORT DECEMBER 2013 1
Tall Man Lettering List REPORT DECEMBER 2013 1 TALL MAN LETTERING LIST REPORT WWW.HQSC.GOVT.NZ Published in December 2013 by the Health Quality & Safety Commission. This document is available on the Health Quality & Safety Commission website, www.hqsc.govt.nz ISBN: 978-0-478-38555-7 (online) Citation: Health Quality & Safety Commission. 2013. Tall Man Lettering List Report. Wellington: Health Quality & Safety Commission. Crown copyright ©. This copyright work is licensed under the Creative Commons Attribution-No Derivative Works 3.0 New Zealand licence. In essence, you are free to copy and distribute the work (including other media and formats), as long as you attribute the work to the Health Quality & Safety Commission. The work must not be adapted and other licence terms must be abided. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nd/3.0/nz/ Copyright enquiries If you are in doubt as to whether a proposed use is covered by this licence, please contact: National Medication Safety Programme Team Health Quality & Safety Commission PO Box 25496 Wellington 6146 ACKNOWLEDGEMENTS The Health Quality & Safety Commission acknowledges the following for their assistance in producing the New Zealand Tall Man lettering list: • The Australian Commission on Safety and Quality in Health Care for advice and support in allowing its original work to be either reproduced in whole or altered in part for New Zealand as per its copyright1 • The Medication Safety and Quality Program of Clinical Excellence Commission, New South -

United States Patent (19) (11) 4,310,524 Wiech Et Al
United States Patent (19) (11) 4,310,524 Wiech et al. 45 Jan. 12, 1982 (54) TCA COMPOSITION AND METHOD FOR McMillen et al., Fed. Proc., 38,592 (1979). RAPD ONSET ANTDEPRESSANT Sellinger et al., Fed. Proc., 38,592 (1979). THERAPY Pandey et al., Fed. Proc., 38,592 (1979). 75) Inventors: Norbert L. Wiech; Richard C. Ursillo, Primary Examiner-Stanley J. Friedman both of Cincinnati, Ohio Attorney, Agent, or Firm-Millen & White 73) Assignee: Richardson-Merrell, Inc., Wilton, Conn. (57 ABSTRACT A method is provided for treating depression in a pa (21) Appl. No.: 139,498 tient therefrom and requiring rapid symptomatic relief, (22 Filed: Apr. 11, 1980 which comprises administering to said patient concur 51) Int. Cl. .................... A61K 31/33; A61K 31/135 rently (a) an effective antidepressant amount of a tricy clic antidepressant or a pharmaceutically effective acid (52) ...... 424/244; 424/330 addition salt thereof, and (b) an amount of an a-adrener 58) Field of Search ................................ 424/244, 330 gic receptor blocking agent effective to achieve rapid (56) References Cited onset of the antidepressant action of (a), whereby the PUBLICATIONS onset of said antidepressant action is achieved within Chemical Abst., vol. 66-72828m, (1967), Kellett. from 1 to 7 days. Chemical Abst, vol. 68-94371a, (1968), Martelli et al. A pharmaceutical composition is also provided which is Chemical Abst., vol. 74-86.048j, (1971), Dixit et al. especially adapted for use with the foregoing method. Holmberg et al., Psychopharm., 2,93 (1961). Svensson, Symp. Med. Hoechst., 13, 245 (1978). 17 Claims, No Drawings 4,310,524 1. -

Package Leaflet: Information for the User Hydroxyzine Bluefish 25 Mg
Package leaflet: Information for the user Hydroxyzine Bluefish 25 mg film-coated tablets hydroxyzine hydrochloride Read all of this leaflet carefully before you start using this medicine because it contains important information for you. - Keep this leaflet. You may need to read it again. - If you have any further questions, ask your doctor or pharmacist. - This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. - If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What Hydroxyzine Bluefish is and what it is used for 2. What you need to know before you use Hydroxyzine Bluefish 3. How to use Hydroxyzine Bluefish 4. Possible side effects 5. How to store Hydroxyzine Bluefish 6. Contents of the pack and other information 1. What Hydroxyzine Bluefish is and what it is used for Hydroxyzine Bluefish blocks histamine, a substance found in body tissues. It is effective against anxiety, itching and urticaria. Hydroxyzine Bluefish is used to treat anxiety in adults itching (pruritus) associated with hives (urticaria) in adults adolescents and children of 5 years and above 2. What you need to know before you take Hydroxyzine Bluefish Do not take Hydroxyzine Bluefish - if you are allergic to hydroxyzine hydrochloride or any of the other ingredients of this medicine (listed in section 6) - if you are allergic (hypersensitive) to cetirizine, aminophylline, ethylenediamine or piperazine derivatives (closely related active substances of other medicines). -

In Vitro Pharmacology of Clinically Used Central Nervous System-Active Drugs As Inverse H1 Receptor Agonists
0022-3565/07/3221-172–179$20.00 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 322, No. 1 Copyright © 2007 by The American Society for Pharmacology and Experimental Therapeutics 118869/3215703 JPET 322:172–179, 2007 Printed in U.S.A. In Vitro Pharmacology of Clinically Used Central Nervous System-Active Drugs as Inverse H1 Receptor Agonists R. A. Bakker,1 M. W. Nicholas,2 T. T. Smith, E. S. Burstein, U. Hacksell, H. Timmerman, R. Leurs, M. R. Brann, and D. M. Weiner Department of Medicinal Chemistry, Leiden/Amsterdam Center for Drug Research, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands (R.A.B., H.T., R.L.); ACADIA Pharmaceuticals Inc., San Diego, California (R.A.B., M.W.N., T.T.S., E.S.B., U.H., M.R.B., D.M.W.); and Departments of Pharmacology (M.R.B.), Neurosciences (D.M.W.), and Psychiatry (D.M.W.), University of California, San Diego, California Received January 2, 2007; accepted March 30, 2007 Downloaded from ABSTRACT The human histamine H1 receptor (H1R) is a prototypical G on this screen, we have reported on the identification of 8R- protein-coupled receptor and an important, well characterized lisuride as a potent stereospecific partial H1R agonist (Mol target for the development of antagonists to treat allergic con- Pharmacol 65:538–549, 2004). In contrast, herein we report on jpet.aspetjournals.org ditions. Many neuropsychiatric drugs are also known to po- a large number of varied clinical and chemical classes of drugs tently antagonize this receptor, underlying aspects of their side that are active in the central nervous system that display potent effect profiles. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0115729 A1 Qin Et Al
US 201201.15729A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0115729 A1 Qin et al. (43) Pub. Date: May 10, 2012 (54) PROCESS FOR FORMING FILMS, FIBERS, Publication Classification AND BEADS FROM CHITNOUS BOMASS (51) Int. Cl (75) Inventors: Ying Qin, Tuscaloosa, AL (US); AOIN 25/00 (2006.01) Robin D. Rogers, Tuscaloosa, AL A6II 47/36 (2006.01) AL(US); (US) Daniel T. Daly, Tuscaloosa, tish 9.8 (2006.01)C (52) U.S. Cl. ............ 504/358:536/20: 514/777; 426/658 (73) Assignee: THE BOARD OF TRUSTEES OF THE UNIVERSITY OF 57 ABSTRACT ALABAMA, Tuscaloosa, AL (US) (57) Disclosed is a process for forming films, fibers, and beads (21) Appl. No.: 13/375,245 comprising a chitinous mass, for example, chitin, chitosan obtained from one or more biomasses. The disclosed process (22) PCT Filed: Jun. 1, 2010 can be used to prepare films, fibers, and beads comprising only polymers, i.e., chitin, obtained from a suitable biomass, (86). PCT No.: PCT/US 10/36904 or the films, fibers, and beads can comprise a mixture of polymers obtained from a suitable biomass and a naturally S3712). (4) (c)(1), Date: Jan. 26, 2012 occurring and/or synthetic polymer. Disclosed herein are the (2), (4) Date: an. AO. films, fibers, and beads obtained from the disclosed process. O O This Abstract is presented solely to aid in searching the sub Related U.S. Application Data ject matter disclosed herein and is not intended to define, (60)60) Provisional applicationpp No. 61/182,833,sy- - - s filed on Jun.