Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using Powerseq Chemistry on the Miseq Fgx System
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ethical Dimensions of NGS Technologies to Criminal Investigations
ETHICAL DIMENSIONS OF THE APPLICATION OF NEXT GENERATION SEQUENCING TECHNOLOGIES TO CRIMINAL INVESTIGATIONS Author: National DNA Database Ethics Group Date: March 2017 1.0 Introduction 1.1 Next Generation Sequencing (NGS) is a term used to describe DNA sequencing technologies whereby multiple pieces of DNA are sequenced in parallel. This allows large sections of the human genome to be sequenced rapidly. The name is a catch- all-phrase that refers to high-throughput sequencing rather than the previous Sanger sequencing technology, which was much slower. NGS is also known as Massive Parallel Sequencing and the terms are often used interchangeably. Within this document the term NGS refers to technologies that provide more wide-ranging information than the standard DNA short tandem repeat (STR) profiling techniques that measure the number of repeats at a specific region of non-coding DNA within an autosomal chromosome. 1.2 NGS sequencing technologies have developed rapidly over the past decade while the costs associated with sequencing have declined. Whilst need and utility, and not merely the availability and affordability of NGS technologies, should be the driver for their introduction into criminal investigations, declining costs increase the feasibility of their introduction. It is therefore timely that the ethical issues associated with the application of NGS in criminal investigations are considered. In this document, the Ethics Group (EG) provides an outline of the NGS technologies that are likely to become available in the next 10 years and a map (albeit not yet an in-depth discussion) of the ethical challenges associated with the application of these technologies for forensic purposes. -
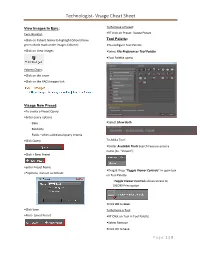
Technologist- Visage Cheat Sheet
Technologist- Visage Cheat Sheet View Images In Epic: To Remove a Preset: Tech Worklist: •RT click on Preset- Delete Preset •Click on Patient Name to highlight (Should have Tool Palette: green check mark under Images Column) •To configure Tool Palette •Click on View Images •Select File-Preferences-Tool Palette •Tool Palette opens Patient Chart: •Click on the exam •Click on the PACS Images link. Visage New Preset: •To create a Preset Query •Enter query options ◦Date •Select Show Both ◦Modality ◦Fields –offers additional query criteria •Click Query To Add a Tool: •Under Available Tools Search Feature-enter a name (Ex. “Viewer”) •Click + New Preset •Enter Preset Name •Drag & Drop “Toggle Viewer Controls” in open box •*Options- Can set as Default on Tool Palette ◦Toggle Viewer Controls allows access to DICOM Print option •Click OK to Save •Click Save To Remove a Tool: •Note- Saved Preset •RT Click on Tool in Tool Palette •Select Remove •Click OK to Save P a g e 1 | 3 Technologist- Visage Cheat Sheet Mark Images for Removal: DICOM Send: •Pull the image into the Active Viewer •Search for patient via the Visage Study Browser •RT click on image & choose Permanent Text Label ◦Name, MRN#, ACC# from Tool Palette *This is Permanent •Click on Accession # to send entire exam •Click the + beside the patient name to open series •Click on the Series that need sent •Draw a box over the image ◦To select multiple series, hold the Ctrl key •Type in DELETE in pop-up box •Change font size to a larger size •Click on Save Modified Images •Click Save •Click Save -
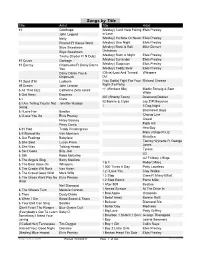
Songs by Title
Songs by Title Title Artist Title Artist #1 Goldfrapp (Medley) Can't Help Falling Elvis Presley John Legend In Love Nelly (Medley) It's Now Or Never Elvis Presley Pharrell Ft Kanye West (Medley) One Night Elvis Presley Skye Sweetnam (Medley) Rock & Roll Mike Denver Skye Sweetnam Christmas Tinchy Stryder Ft N Dubz (Medley) Such A Night Elvis Presley #1 Crush Garbage (Medley) Surrender Elvis Presley #1 Enemy Chipmunks Ft Daisy Dares (Medley) Suspicion Elvis Presley You (Medley) Teddy Bear Elvis Presley Daisy Dares You & (Olivia) Lost And Turned Whispers Chipmunk Out #1 Spot (TH) Ludacris (You Gotta) Fight For Your Richard Cheese #9 Dream John Lennon Right (To Party) & All That Jazz Catherine Zeta Jones +1 (Workout Mix) Martin Solveig & Sam White & Get Away Esquires 007 (Shanty Town) Desmond Dekker & I Ciara 03 Bonnie & Clyde Jay Z Ft Beyonce & I Am Telling You Im Not Jennifer Hudson Going 1 3 Dog Night & I Love Her Beatles Backstreet Boys & I Love You So Elvis Presley Chorus Line Hirley Bassey Creed Perry Como Faith Hill & If I Had Teddy Pendergrass HearSay & It Stoned Me Van Morrison Mary J Blige Ft U2 & Our Feelings Babyface Metallica & She Said Lucas Prata Tammy Wynette Ft George Jones & She Was Talking Heads Tyrese & So It Goes Billy Joel U2 & Still Reba McEntire U2 Ft Mary J Blige & The Angels Sing Barry Manilow 1 & 1 Robert Miles & The Beat Goes On Whispers 1 000 Times A Day Patty Loveless & The Cradle Will Rock Van Halen 1 2 I Love You Clay Walker & The Crowd Goes Wild Mark Wills 1 2 Step Ciara Ft Missy Elliott & The Grass Wont Pay -

Songs by Title
Karaoke Song Book Songs by Title Title Artist Title Artist #1 Nelly 18 And Life Skid Row #1 Crush Garbage 18 'til I Die Adams, Bryan #Dream Lennon, John 18 Yellow Roses Darin, Bobby (doo Wop) That Thing Parody 19 2000 Gorillaz (I Hate) Everything About You Three Days Grace 19 2000 Gorrilaz (I Would Do) Anything For Love Meatloaf 19 Somethin' Mark Wills (If You're Not In It For Love) I'm Outta Here Twain, Shania 19 Somethin' Wills, Mark (I'm Not Your) Steppin' Stone Monkees, The 19 SOMETHING WILLS,MARK (Now & Then) There's A Fool Such As I Presley, Elvis 192000 Gorillaz (Our Love) Don't Throw It All Away Andy Gibb 1969 Stegall, Keith (Sitting On The) Dock Of The Bay Redding, Otis 1979 Smashing Pumpkins (Theme From) The Monkees Monkees, The 1982 Randy Travis (you Drive Me) Crazy Britney Spears 1982 Travis, Randy (Your Love Has Lifted Me) Higher And Higher Coolidge, Rita 1985 BOWLING FOR SOUP 03 Bonnie & Clyde Jay Z & Beyonce 1985 Bowling For Soup 03 Bonnie & Clyde Jay Z & Beyonce Knowles 1985 BOWLING FOR SOUP '03 Bonnie & Clyde Jay Z & Beyonce Knowles 1985 Bowling For Soup 03 Bonnie And Clyde Jay Z & Beyonce 1999 Prince 1 2 3 Estefan, Gloria 1999 Prince & Revolution 1 Thing Amerie 1999 Wilkinsons, The 1, 2, 3, 4, Sumpin' New Coolio 19Th Nervous Breakdown Rolling Stones, The 1,2 STEP CIARA & M. ELLIOTT 2 Become 1 Jewel 10 Days Late Third Eye Blind 2 Become 1 Spice Girls 10 Min Sorry We've Stopped Taking Requests 2 Become 1 Spice Girls, The 10 Min The Karaoke Show Is Over 2 Become One SPICE GIRLS 10 Min Welcome To Karaoke Show 2 Faced Louise 10 Out Of 10 Louchie Lou 2 Find U Jewel 10 Rounds With Jose Cuervo Byrd, Tracy 2 For The Show Trooper 10 Seconds Down Sugar Ray 2 Legit 2 Quit Hammer, M.C. -

Evaluation of Openarray™ As a Genotyping Method for Forensic DNA Phenotyping and Human Identification
G C A T T A C G G C A T genes Article Evaluation of OpenArray™ as a Genotyping Method for Forensic DNA Phenotyping and Human Identification Michele Ragazzo 1, Giulio Puleri 1, Valeria Errichiello 1, Laura Manzo 1, Laura Luzzi 1 , Saverio Potenza 2 , Claudia Strafella 1,3 , Cristina Peconi 3, Fabio Nicastro 4 , Valerio Caputo 1 and Emiliano Giardina 1,3,* 1 Department of Biomedicine and Prevention, Tor Vergata University of Rome, 00133 Rome, Italy; [email protected] (M.R.); [email protected] (G.P.); [email protected] (V.E.); [email protected] (L.M.); [email protected] (L.L.); [email protected] (C.S.); [email protected] (V.C.) 2 Department of Biomedicine and Prevention, Section of Legal Medicine, Social Security and Forensic Toxicology, University of Rome Tor Vergata, 00133 Rome, Italy; [email protected] 3 Genomic Medicine Laboratory UILDM, IRCCS Santa Lucia Foundation, 00179 Rome, Italy; [email protected] 4 Austech, 00153 Rome, Italy; [email protected] * Correspondence: [email protected] Abstract: A custom plate of OpenArray™ technology was evaluated to test 60 single-nucleotide polymorphisms (SNPs) validated for the prediction of eye color, hair color, and skin pigmentation, and for personal identification. The SNPs were selected from already validated subsets (Hirisplex-s, Precision ID Identity SNP Panel, and ForenSeq DNA Signature Prep Kit). The concordance rate and call rate for every SNP were calculated by analyzing 314 sequenced DNA samples. The sensitivity of the assay was assessed by preparing a dilution series of 10.0, 5.0, 1.0, and 0.5 ng. -

About the AAFS
American Academy of Forensic Sciences 410 North 21st Street Colorado Springs, Colorado 80904 Phone: (719) 636-1100 Email: [email protected] Website: www.aafs.org @ AAFS Publication 20-2 Copyright © 2020 American Academy of Forensic Sciences Printed in the United States of America Publication Printers, Inc., Denver, CO Typography by Kathy Howard Cover Art by My Creative Condition, Colorado Springs, CO WELCOME LETTER Dear Attendees, It is my high honor and distinct privilege to welcome you to the 72nd AAFS Annual Scientific Meeting in Anaheim, California. I would like to thank the AAFS staff, the many volunteers, and everyone else who have worked together to create an excellent program for this meeting with the theme Crossing Borders. You will have many opportunities to meet your colleagues and discuss new challenges in the field. There are many workshops and special sessions that will be presented. The Interdisciplinary and Plenary Sessions will provide different views in forensic science—past, present, and future. The Young Forensic Scientists Forum will celebrate its 25th Anniversary and is conducting a workshop related to the meeting theme. More than 1,000 presentations are scheduled that will provide you with more insight into the developments in forensic science. The exhibit hall, always interesting to explore, is where you will see the latest forensic science equipment, technology, and literature. The theme Crossing Borders was chosen by me and my colleagues at the Netherlands Forensic Institute (NFI). We see many definitions of crossing borders in forensic science today. For the 2020 meeting, six words starting with the letters “IN” are included in the theme. -

Top 40 Singles Top 40 Albums
21 June 1981 CHART #288 Top 40 Singles Top 40 Albums Love Will Tear Us Apart The Bridge Unknown Pleasures Vienna 1 Joy Division 21 Deane Waretini 1 Joy Division 21 Ultravox Last week 41 / 2 weeks RTC Last week 19 / 20 weeks Gold / CBS Last week - / 1 weeks Gold / RTC Last week 31 / 20 weeks FESTIVAL Stars On 45 Boom Boom Faith Another Ticket 2 Stars on 45 22 Black Slate 2 The Cure 22 Eric Clapton Last week 5 / 3 weeks POLYGRAM Last week 16 / 11 weeks POLYGRAM Last week 1 / 4 weeks Gold / CBS Last week - / 1 weeks POLYGRAM Bette Davis Eyes Baggy Trousers Ep Sucking In The Seventies Zebop! 3 Kim Carnes 23 Madness 3 Rolling Stones 23 Santana Last week 3 / 5 weeks EMI Last week 28 / 7 weeks POLYGRAM Last week 11 / 2 weeks Gold / EMI Last week 23 / 2 weeks CBS (Morning Train) Nine To Five Just The Two Of Us Reveries Journeys To Glory 4 Sheena Easton 24 Grover Washington Jnr 4 Richard Clayderman 24 Spandau Ballet Last week 1 / 6 weeks EMI Last week 39 / 3 weeks WEA Last week 5 / 13 weeks Platinum / WEA Last week 20 / 3 weeks FESTIVAL This Ole House Fade To Grey Waiata Hotter Than July 5 Shakin' Stevens 25 Visage 5 Split Enz 25 Stevie Wonder Last week 4 / 6 weeks CBS Last week 10 / 12 weeks UNIVERSAL Last week 2 / 9 weeks Platinum / POLYGRAM Last week 16 / 27 weeks Platinum / EMI How 'Bout Us Ant Music Being With You Shades 6 Champaign 26 Adam and the Ants 6 Smokey Robinson 26 JJ Cale Last week 6 / 5 weeks CBS Last week 21 / 17 weeks CBS Last week 7 / 6 weeks EMI Last week 21 / 6 weeks FESTIVAL On The Inside Blam Blam Blam EP Making Movies Seventeen -

Forensic DNA Phenotyping for Degraded Samples
Forensic DNA Phenotyping for Degraded Samples Ronak Hassani Nejad, Steen Harsten, and Jason Moore British Colombia Institution of Technology 9. A threshold of higher or equal to 0.7 was considered when deter- Abstract mining the final eye color predictions. 10. For hair color predictions, the highest p-value among the 4 color Forensic DNA phenotyping (FDP) is a novel technique that can assist category and the shade color was considered in final determina- the investigation by predicting a person of interest’s external visible tion of hair color. characteristics (EVCs) and phenotypes such as eye, hair, and skin col- our (Walsh, 2013). Several DNA markers with the prediction ability Results for these EVCs are recognized and various commercial and non- commercial tools were developed for practical use in forensic labora- The majority of samples had a p-value above 0.7 for all of the tests of tories. the experiment in eye colors even at 16% (22 samples out of 30) (Figure 3). This study investigates the compatibility of the Verogen Universal Analysis System (UAS) with the HIrisPlex Webtool. This study also tests the HIrisPlex Webtool’s reliability to generate accurate pheno- typing predictions for degraded samples through a series of tests in which we intentionally created degraded samples by deleting SNP data from the DNA profile. For p-values resulted for brown and blond hair color categories, there is a noticeable fluctuation between the tests (Figure 8). This fluctuation indicates that some individual SNPs have more predic- Introduction tive power than others. Eye color prediction results had less predic- tion fluctuations (figure 7) than hair colour. -

Phenotype Report Case #91-2200
Snapshot Prediction Results Phenotype Report Case #91-2200 Contact: Lt. Michael Buffington League City Police Dept. (281) 338-8220 Sex: Female♀ Age: Unknown (Shown at age 25) Body Mass: Unknown (Shown at BMI 22, Normal) Ancestry: Western European Likely Family Origin: Louisiana 10.9 NOT: Brown / Dark Brown Skin Color (97.8% confidence) Fair / Light Brown (74.2% confidence) 68.4 NOT: Blue / Black Eye Color (96.9% confidence) Hazel / Green (86.2% confidence) 28.7 NOT: Red / Black Hair Color (92.1% confidence) Brown / Blond (92.1% confidence) Freckles 40.0 Zero / Few (70.2% confidence) © 2018 Parabon NanoLabs, Inc. All rights reserved. https://Parabon-NanoLabs.com/Snapshot Investigators Seek Public’s Help Identifying a Homicide Victim With Ties to Louisiana For Immediate Release: League City Police Use New DNA Methods in Attempt to Identify 1991 Female Murder Victim League City, Texas – Investigators from the League City Police Department are seeking the public’s help in identifying the skeletal remains of a homicide victim from a 1991 “Calder Road” cold case. Detectives are hoping that recent scientific advancements in the field of forensic DNA analysis will generate leads and help identify the woman whose body was found on September 8, 1991, in League City, Galveston County, Texas. League City investigators recently employed Parabon NanoLabs (Parabon), a DNA technology company in Virginia, to predict the physical appearance and ancestry of the unidentified woman using a new method of forensic DNA analysis called DNA phenotyping. Parabon’s Snapshot® DNA Phenotyping software analyzed the unidentified woman’s DNA and made predictions about her eye color, hair color, skin color, freckling, and face shape. -

DNA Phenotyping and Kinship Determination
DNA Phenotyping and Kinship Determination Ellen McRae Greytak, PhD Director of Bioinformatics Parabon NanoLabs, Inc. ©©2 2015015 ParabonParabon NanoLabs,NanoLabs Inc.Inc All rightsrights reserved. reserved Forensic Applications of DNA Phenotyping Predict a person’s ancestry and/or appearance (“phenotype”) from his or her DNA Generate investigative leads when DNA doesn’t match a database (e.g., CODIS) Gain additional information (e.g., pigmentation, detailed ancestry) about unidentified remains Main value is in excluding non-matching individuals to help narrow a suspect list Without information on age, weight, lifestyle, etc., phenotyping currently is not targeted toward individual identification Snapshot Workflow Workflow of a Parabon® Snapshot™ Investigation Unidentified Remains DNA Evidence Is Collected and Sent to Crime Lab DNA Evidence DNA Crime Lab CCrime Lab Extracts DNA And Produces STR Profile Checked STR Profile (a.k.a. “DNA Fingerprint”) AAgainst DNA Database(s) Yes Match No Found? SnapshotS Composite Ordered Extracted DNA ™ D N A PH E N O T Y P I N G DNA Service Labs Unidentified DNA Is Genotype Data Is Genotyping Lab Produces SNP Sent To Service Lab Sent To Parabon Profile (a.k.a. “DNA Blueprint”) (DNA Extracted If Needed) 50pg – 2ng DNA Evidence — or — Extracted DNA NOTE: STR Profiles Do Not Contain Sufficient Genetic Information to Produce A SNP Genotype Parabon NanoLabs PParabonb AnalyzesAl PParabon Predicts Physical Traits Investigator Uses Genotype Data and Produces Snapshot Report -

FORENSIC DNA PHENOTYPING and MASSIVE PARALLEL SEQUENCING by Krystal Breslin
FORENSIC DNA PHENOTYPING AND MASSIVE PARALLEL SEQUENCING by Krystal Breslin A Thesis Submitted to the Faculty of Purdue University In Partial Fulfillment of the Requirements for the degree of Master of Science Department of Biological Sciences Indianapolis, Indiana December 2017 ii THE PURDUE UNIVERSITY GRADUATE SCHOOL STATEMENT OF COMMITTEE APPROVAL Dr. Susan Walsh, Chair Department of Biology Dr. Kathleen Marrs Department of Biology Dr. Benjamin Perrin Department of Biology Approved by: Dr. Stephen Randall Head of the Graduate Program iii Dedicated to my husband and two sons. Thank you for always giving me the courage to follow my dreams, no matter where they take us. iv ACKNOWLEDGMENTS I would like to acknowledge and thank everyone who has helped me to become the person I am today. I would like to begin by thanking Dr. Susan Walsh, my PI and mentor. Your passion for science is contagious and has been instrumental in helping to inspire me to pursue my goals. Without your trust and guidance, I would not have been able to grow into the scientist I am today. You are a true role model and I am eternally grateful to have worked with you for the last three years. I cannot wait to see where the future takes you and your research. Secondly, I would like to thank Dr. Ben Perrin and Dr. Kathy Marrs for being a valuable part of my advisory committee. You are both very impressive scientists, and I truly value the input you have provided to my thesis. I would also like to acknowledge and thank each and every member of the Walsh laboratory: Charanya, Ryan, Noah, Morgan, Mirna, Bailey, Stephanie, Lydia, Emma, Clare, Sarah, Megan, Annie, Wesli, Kirsten, Katherine, and Gina. -

Analysis of Electroacoustic Works with Music and Language Intersections
ANALYSIS OF ELECTROACOUSTIC WORKS WITH MUSIC AND LANGUAGE INTERSECTIONS Rodrigo Segnini Bruno Ruviaro Center for Computer Research Center for Computer Research in Music and Acoustics (CCRMA) in Music and Acoustics (CCRMA) Stanford University Stanford University ABSTRACT 2. METHODOLOGY We have selected 15 pieces that invite fundamental This paper proposes a theoretical model for the analysis questions about the boundaries between music and of music and language intersections in contemporary speech. Each piece meets the following criteria: music. This model is tested on a repertoire of • Vocal sounds in any imaginable form should play a electroacoustic and acoustic pieces, providing examples relevant role in it. Synthetic sounds that emulate of blurred boundaries between music and speech. We speech or other vocal behaviors also qualify. have defined two axes representing possible dimensions • It should be representative of the various post-tonal of this relationship: intelligibility and music/speech musical structuring processes that flourished qualities. The resulting sonic space provides a visual throughout the 20th century. account that can serve as a “travel guide” in the music- speech continuum. We envision applications of this Composer Piece * model in the fields of composition, musicology, and SCHOENBERG, Arnold Pierrot Lunaire (1912) 1v + cognitive studies. (1874-1951) ins SCHWITTERS, Kurt Ursonate (1922-32) 1v (1887-1948) 1. INTRODUCTION SCHAEFFER, Pierre (1910- Symphonie pour ea The human voice has played a significant role in