Cross-Species, Amplifiable EST-SSR Markers for Amentotaxus Species
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spatial Distribution and Historical Dynamics of Threatened Conifers of the Dalat Plateau, Vietnam
SPATIAL DISTRIBUTION AND HISTORICAL DYNAMICS OF THREATENED CONIFERS OF THE DALAT PLATEAU, VIETNAM A thesis Presented to The Faculty of the Graduate School At the University of Missouri In Partial Fulfillment Of the Requirements for the Degree Master of Arts By TRANG THI THU TRAN Dr. C. Mark Cowell, Thesis Supervisor MAY 2011 The undersigned, appointed by the dean of the Graduate School, have examined the thesis entitled SPATIAL DISTRIBUTION AND HISTORICAL DYNAMICS OF THREATENED CONIFERS OF THE DALAT PLATEAU, VIETNAM Presented by Trang Thi Thu Tran A candidate for the degree of Master of Arts of Geography And hereby certify that, in their opinion, it is worthy of acceptance. Professor C. Mark Cowell Professor Cuizhen (Susan) Wang Professor Mark Morgan ACKNOWLEDGEMENTS This research project would not have been possible without the support of many people. The author wishes to express gratitude to her supervisor, Prof. Dr. Mark Cowell who was abundantly helpful and offered invaluable assistance, support, and guidance. My heartfelt thanks also go to the members of supervisory committees, Assoc. Prof. Dr. Cuizhen (Susan) Wang and Prof. Mark Morgan without their knowledge and assistance this study would not have been successful. I also wish to thank the staff of the Vietnam Initiatives Group, particularly to Prof. Joseph Hobbs, Prof. Jerry Nelson, and Sang S. Kim for their encouragement and support through the duration of my studies. I also extend thanks to the Conservation Leadership Programme (aka BP Conservation Programme) and Rufford Small Grands for their financial support for the field work. Deepest gratitude is also due to Sub-Institute of Ecology Resources and Environmental Studies (SIERES) of the Institute of Tropical Biology (ITB) Vietnam, particularly to Prof. -

Gymnosperms) of New York State
QK 129 . C667 1992 Pinophyta (Gymnosperms) of New York State Edward A. Cope The L. H. Bailey Hortorium Cornell University Contributions to a Flora of New York State IX Richard S. Mitchell, Editor 1992 Bulletin No. 483 New York State Museum The University of the State of New York THE STATE EDUCATION DEPARTMENT Albany, New York 12230 V A ThL U: ESTHER T. SVIERTZ LIBRARY THI-: ?‘HW YORK BOTANICAL GARDEN THE LuESTHER T. MERTZ LIBRARY THE NEW YORK BOTANICAL GARDEN Pinophyta (Gymnosperms) of New York State Edward A. Cope The L. H. Bailey Hortorium Cornell University Contributions to a Flora of New York State IX Richard S. Mitchell, Editor 1992 Bulletin No. 483 New York State Museum The University of the State of New York THE STATE EDUC ATION DEPARTMENT Albany, New York 12230 THE UNIVERSITY OF THE STATE OF NEW YORK Regents of The University Martin C. Barell, Chancellor, B.A., I.A., LL.B. Muttontown R. Carlos Carballada, Vice Chancellor, B.S. Rochester Willard A. Genrich, LL.B. Buffalo Emlyn I. Griffith. A.B.. J.D. Rome Jorge L. Batista, B.A.. J.D. Bronx Laura Bradley Chodos, B.A., M.A. Vischer Ferry Louise P. Matteoni, B.A., M.A., Ph.D. Bayside J. Edward Meyer, B.A., LL.B. Chappaqua FloydS. Linton, A.B., M.A., M.P.A. Miller Place Mimi Levin Lif.ber, B.A., M.A. Manhattan Shirley C. Brown, B.A., M.A., Ph.D. Albany Norma Gluck, B.A., M.S.W. Manhattan Adelaide L. Sanford, B.A., M.A., P.D. -

(Pinus Taeda L.) with Related Species
RESEARCH ARTICLE Complete chloroplast genome sequence and comparative analysis of loblolly pine (Pinus taeda L.) with related species Sajjad Asaf1, Abdul Latif Khan1, Muhammad Aaqil Khan2, Raheem Shahzad2, Lubna3, Sang Mo Kang2, Ahmed Al-Harrasi1, Ahmed Al-Rawahi1, In-Jung Lee2,4* 1 Chair of Oman's Medicinal Plants & Marine Natural Products, University of Nizwa, Nizwa, Oman, 2 School of Applied Biosciences, Kyungpook National University, Daegu, Republic of Korea, 3 Department of Botany, Garden Campus, Abdul Wali Khan University Mardan, Mardan, Pakistan, 4 Research Institute for Dok-do and Ulleung-do Island, Kyungpook National University, Daegu, Republic of Korea a1111111111 a1111111111 * [email protected] a1111111111 a1111111111 a1111111111 Abstract Pinaceae, the largest family of conifers, has a diversified organization of chloroplast (cp) genomes with two typical highly reduced inverted repeats (IRs). In the current study, we determined the complete sequence of the cp genome of an economically and ecologically OPEN ACCESS important conifer tree, the loblolly pine (Pinus taeda L.), using Illumina paired-end sequenc- Citation: Asaf S, Khan AL, Khan MA, Shahzad R, ing and compared the sequence with those of other pine species. The results revealed a Lubna , Kang SM, et al. (2018) Complete chloroplast genome sequence and comparative genome size of 121,531 base pairs (bp) containing a pair of 830-bp IR regions, distinguished analysis of loblolly pine (Pinus taeda L.) with by a small single copy (42,258 bp) and large single copy (77,614 bp) region. The chloroplast related species. PLoS ONE 13(3): e0192966. genome of P. taeda encodes 120 genes, comprising 81 protein-coding genes, four ribo- https://doi.org/10.1371/journal.pone.0192966 somal RNA genes, and 35 tRNA genes, with 151 randomly distributed microsatellites. -

Medicinal Plant Conservation
MEDICINAL Medicinal Plant PLANT SPECIALIST GROUP Conservation Silphion Volume 11 Newsletter of the Medicinal Plant Specialist Group of the IUCN Species Survival Commission Chaired by Danna J. Leaman Chair’s note . 2 Sustainable sourcing of Arnica montana in the International Standard for Sustainable Wild Col- Apuseni Mountains (Romania): A field project lection of Medicinal and Aromatic Plants – Wolfgang Kathe . 27 (ISSC-MAP) – Danna Leaman . 4 Rhodiola rosea L., from wild collection to field production – Bertalan Galambosi . 31 Regional File Conservation data sheet Ginseng – Dagmar Iracambi Medicinal Plants Project in Minas Gerais Lange . 35 (Brazil) and the International Standard for Sus- tainable Wild Collection of Medicinal and Aro- Conferences and Meetings matic Plants (ISSC-MAP) – Eleanor Coming up – Natalie Hofbauer. 38 Gallia & Karen Franz . 6 CITES News – Uwe Schippmann . 38 Conservation aspects of Aconitum species in the Himalayas with special reference to Uttaran- Recent Events chal (India) – Niranjan Chandra Shah . 9 Conservation Assessment and Management Prior- Promoting the cultivation of medicinal plants in itisation (CAMP) for wild medicinal plants of Uttaranchal, India – Ghayur Alam & Petra North-East India – D.K. Ved, G.A. Kinhal, K. van de Kop . 15 Ravikumar, R. Vijaya Sankar & K. Haridasan . 40 Taxon File Notices of Publication . 45 Trade in East African Aloes – Sara Oldfield . 19 Towards a standardization of biological sustain- List of Members. 48 ability: Wildcrafting Rhatany (Krameria lap- pacea) in Peru – Maximilian -

PHYLOGENETIC RELATIONSHIPS of TORREYA (TAXACEAE) INFERRED from SEQUENCES of NUCLEAR RIBOSOMAL DNA ITS REGION Author(S): Jianhua Li, Charles C
PHYLOGENETIC RELATIONSHIPS OF TORREYA (TAXACEAE) INFERRED FROM SEQUENCES OF NUCLEAR RIBOSOMAL DNA ITS REGION Author(s): Jianhua Li, Charles C. Davis, Michael J. Donoghue, Susan Kelley and Peter Del Tredici Source: Harvard Papers in Botany, Vol. 6, No. 1 (July 2001), pp. 275-281 Published by: Harvard University Herbaria Stable URL: http://www.jstor.org/stable/41761652 Accessed: 14-06-2016 15:35 UTC REFERENCES Linked references are available on JSTOR for this article: http://www.jstor.org/stable/41761652?seq=1&cid=pdf-reference#references_tab_contents You may need to log in to JSTOR to access the linked references. Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at http://about.jstor.org/terms JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. Harvard University Herbaria is collaborating with JSTOR to digitize, preserve and extend access to Harvard Papers in Botany This content downloaded from 128.103.224.4 on Tue, 14 Jun 2016 15:35:14 UTC All use subject to http://about.jstor.org/terms PHYLOGENETIC RELATIONSHIPS OF TORREYA (TAXACEAE) INFERRED FROM SEQUENCES OF NUCLEAR RIBOSOMAL DNA ITS REGION Jianhua Li,1 Charles C. Davis,2 Michael J. Donoghue,3 Susan Kelley,1 And Peter Del Tredici1 Abstract. Torreya, composed of five to seven species, is distributed disjunctly in eastern Asia and the eastern and western United States. -

Vietnamese Conifers and Some Problems of Their Sustainable Utilization Ke Loc Et Al
Vietnamese conifers and some problems of their sustainable utilization Ke Loc et al. Vietnamese conifers and some problems of their sustainable utilization Phan Ke Loc 1, 2, Nguyen Tien Hiep 2, Nguyen Duc To Luu 3, Philip Ian Thomas 4, Aljos Farjon 5, L.V. Averyanov 6, J.C. Regalado, Jr. 7, Nguyen Sinh Khang 2, Georgina Magin 8, Paul Mathew 8, Sara Oldfield 9, Sheelagh O’Reilly 8, Thomas Osborn 10, Steven Swan 8 and To Van Thao 2 1 University of Natural Science, Vietnam National University, Hanoi; 2 Institute of Ecology and Biological Resources; 3 Vietnam Central Forest Seed Company; 4 Royal Botanic Garden Edinburgh; 5 Royal Botanic Gardens, Kew; 6 Komarov Botanical Institute; 7 Missouri Botanical Garden; 8 Fauna & Flora International; 9 Global Trees Campaign; 10 Independent Consultant Introduction Vietnam is now recognized as one of the top ten global conifer conservation ‘hotspots’, as defined by the Conifer Specialist Group of the World Conservation Union (IUCN). Vietnam’s conifer flora has approximately 34 species that are indigenous to the country, making up about 5% of conifers known worldwide. Although conifers represent only less than 0.3% of the total number of higher vascular plant species of Vietnam, they are of great ecological, cultural and economic importance. Most conifer wood is prized for its high value in house construction, furniture making, etc. The decline of conifer populations in Vietnam has caused serious concern among scientists. Threats to conifer species are substantial and varied, ranging from logging (both commercial and subsistence), land clearing for agriculture, and forest fire. Over the past twelve years (1995-2006), Vietnam Botanical Conservation Program (VBCP), a scientific cooperation between the Missouri Botanical Garden in Saint Louis and the Institute of Ecology and Biological Resources in Hanoi, has conducted various studies on this important group of plants in order to gather baseline information necessary to make sound recommendations for their conservation and sustainable use. -

Mcabee Fossil Site Assessment
1 McAbee Fossil Site Assessment Final Report July 30, 2007 Revised August 5, 2007 Further revised October 24, 2008 Contract CCLAL08009 by Mark V. H. Wilson, Ph.D. Edmonton, Alberta, Canada Phone 780 435 6501; email [email protected] 2 Table of Contents Executive Summary ..............................................................................................................................................................3 McAbee Fossil Site Assessment ..........................................................................................................................................4 Introduction .......................................................................................................................................................................4 Geological Context ...........................................................................................................................................................8 Claim Use and Impact ....................................................................................................................................................10 Quality, Abundance, and Importance of the Fossils from McAbee ............................................................................11 Sale and Private Use of Fossils from McAbee..............................................................................................................12 Educational Use of Fossils from McAbee.....................................................................................................................13 -

Number 3, Spring 1998 Director’S Letter
Planning and planting for a better world Friends of the JC Raulston Arboretum Newsletter Number 3, Spring 1998 Director’s Letter Spring greetings from the JC Raulston Arboretum! This garden- ing season is in full swing, and the Arboretum is the place to be. Emergence is the word! Flowers and foliage are emerging every- where. We had a magnificent late winter and early spring. The Cornus mas ‘Spring Glow’ located in the paradise garden was exquisite this year. The bright yellow flowers are bright and persistent, and the Students from a Wake Tech Community College Photography Class find exfoliating bark and attractive habit plenty to photograph on a February day in the Arboretum. make it a winner. It’s no wonder that JC was so excited about this done soon. Make sure you check of themselves than is expected to seedling selection from the field out many of the special gardens in keep things moving forward. I, for nursery. We are looking to propa- the Arboretum. Our volunteer one, am thankful for each and every gate numerous plants this spring in curators are busy planting and one of them. hopes of getting it into the trade. preparing those gardens for The magnolias were looking another season. Many thanks to all Lastly, when you visit the garden I fantastic until we had three days in our volunteers who work so very would challenge you to find the a row of temperatures in the low hard in the garden. It shows! Euscaphis japonicus. We had a twenties. There was plenty of Another reminder — from April to beautiful seven-foot specimen tree damage to open flowers, but the October, on Sunday’s at 2:00 p.m. -

Methods for Measuring Frost Tolerance of Conifers: a Systematic Map
Review Methods for Measuring Frost Tolerance of Conifers: A Systematic Map Anastasia-Ainhoa Atucha Zamkova *, Katherine A. Steele and Andrew R. Smith School of Natural Sciences, Bangor University, Bangor LL57 2UW, Gwynedd, UK; [email protected] (K.A.S.); [email protected] (A.R.S.) * Correspondence: [email protected] Abstract: Frost tolerance is the ability of plants to withstand freezing temperatures without unrecov- erable damage. Measuring frost tolerance involves various steps, each of which will vary depending on the objectives of the study. This systematic map takes an overall view of the literature that uses frost tolerance measuring techniques in gymnosperms, focusing mainly on conifers. Many different techniques have been used for testing, and there has been little change in methodology since 2000. The gold standard remains the field observation study, which, due to its cost, is frequently substituted by other techniques. Closed enclosure freezing tests (all non-field freezing tests) are done using various types of equipment for inducing artificial freezing. An examination of the literature indicates that several factors have to be controlled in order to measure frost tolerance in a manner similar to observation in a field study. Equipment that allows controlling the freezing rate, frost exposure time and thawing rate would obtain results closer to field studies. Other important factors in study design are the number of test temperatures used, the range of temperatures selected and the decrements between the temperatures, which should be selected based on expected frost tolerance of the tissue and species. Citation: Atucha Zamkova, A.-A.; Steele, K.A.; Smith, A.R. -

3. AMENTOTAXUS Pilger, Bot. Jahrb. Syst. 54: 41. 1916
Flora of China 4: 92–94. 1999. 3. AMENTOTAXUS Pilger, Bot. Jahrb. Syst. 54: 41. 1916. 穗花杉属 sui hua shan shu Shrubs or small trees dioecious; branchlets opposite, base with bud scales not persistent; winter buds tetragonal- ovate, acute, glossy, scales, decussate, in 3–5 whorles of 4, ridged adaxially. Leaves decussate, but brought into a single plane by twisting of petioles; blade straight or slightly falcate, usually more than 5 mm wide, adaxial surface mottled when fresh, rarely smooth, rugose or ± so when dry, resin canal present below sheath of vascular bundle, sclereids present, rarely absent, base decurrent, margin slightly downcurved. Pollen cones aggregated into (1 or)2– 6(–10) long, slender, compound racemes or spikes arising from bract axil near apex of branches; individual cones opposite, sessile or subsessile, ellipsoid or subglobose; microsporophylls numerous, ± shield-shaped; pollen sacs 3– 8, arranged radially, or adaxially and abaxially. Seed-bearing structures compressed-tetragonal or flattened abaxially, basal part with 6–10 pairs of decussate bracts arranged in 4 rows each of 3–5 bracts; ovule 1, sessile, erect. Seed ripening in 1st year, long pedunculate, ellipsoid or obovoid-ellipsoid, enclosed except for apex in a saclike aril which is bright red or reddish yellow when ripe; bracts persistent at base. Five or six species: China, Vietnam; three species (one endemic) in China. Amentotaxus has been placed in its own tribe (Amentotaxeae W. C. Cheng & C. D. Chu) or family, Amentotaxaceae. Although Page (in Kubitzki, Fam. Gen. Vasc. Pl. 1: 299–302. 1990) included it in the Cephalotaxaceae, molecular data indicate very strongly that it is the sister genus of Torreya, which is similar in the size and shape of its seed and in usually having bilaterally symmetric clusters of pollen sacs. -

Population Genetic Analysis of Pinus Koraiensis in China Inferred Using EST- SSR Markers
Population Genetic Analysis of Pinus Koraiensis in China Inferred Using EST- SSR Markers Xiang Li State Key Laboratory of Tree Genetics and Breeding, School of Forestry, Northeast Forestry University Minghui Zhao Stat Key Laboratory of Tree Genetics and breeding, School of Forestry, Northeast Forestry University Yujin Xu State Key Laboratory of Tree Genetics and Breeding, School of Forestry, Northeast Forestry University Yan Li State Key Laboratory of Tree Genetics and Breeding, School of Forestry, Northeast Forestry University Mulualem Tigabu Southern Swedish Forest Research Centre, Swedish University of Agricultural Sciences Xiyang Zhao ( [email protected] ) State Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University https://orcid.org/0000-0002-1744-5431 Research article Keywords: Pinus koraiensis, EST-SSRs, genetic diversity, population structure, population differentiation, gene ow, China Posted Date: December 7th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-117881/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract Background Pinus koraiensis (commonly known as Korean pine), is a well-known conifer species in China with high economic, ornamental and ecological values. More than 50% of the P. koraiensis forests in the world are distributed in Northeast China, a region with abundant germplasm resources. However, these natural P. koraiensis populations are in danger of genetic erosion caused by continuous climate changes and frequent human activity. Little work has been conducted on the population genetic structure and genetic differentiation of P. koraiensis in China. Here, representative individuals from 16 natural P. koraiensis populations were sampled and genotyped, and polymorphic expressed sequence tag-simple sequence repeat (EST-SSR) markers were used to comprehensively evaluate genetic diversity, population structure and differentiation of P. -
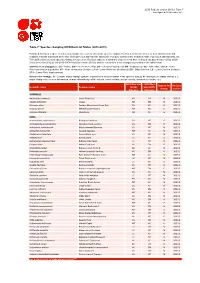
Table 7: Species Changing IUCN Red List Status (2012-2013)
IUCN Red List version 2013.2: Table 7 Last Updated: 25 November 2013 Table 7: Species changing IUCN Red List Status (2012-2013) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2012 (IUCN Red List version 2012.2) and 2013 (IUCN Red List version 2013.2) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered, EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.) IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2012) List (2013) change version Category Category MAMMALS Nycticebus javanicus Javan Slow Loris EN CR N 2013.2 Okapia johnstoni Okapi NT EN N 2013.2 Pteropus niger Greater Mascarene Flying