Identification of Genetic Variants Predictive of Early Onset Pancreatic Cancer Through a Population Science Analysis of Functional Genomic Datasets
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded from NCBI (
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.21.435734; this version posted March 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. Computational modeling of C-terminal tails to predict the calcium-dependent secretion of ER resident proteins Kathleen A. Trychta1,†, Bing Xie2,† , Ravi Kumar Verma2, Min Xu2, Lei Shi2,*, Brandon K. Harvey1,* 1 Molecular Mechanisms of Cellular Stress and Inflammation Unit 2 Computational Chemistry and Molecular Biophysics Section National Institute on Drug Abuse, National Institutes of Health, Baltimore, MD, 21224 † These authors have contributed equally to this work and share first authorship. * These authors share senior authorship. Correspondence: [email protected] [email protected] Key Words: ER calcium, thapsigargin, KDEL receptor, ER retention sequence, exodosis Word Count: 4831 Figures: 4 Supplemental Tables: 2 Supplemental Figures: 3 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.21.435734; this version posted March 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. Abstract The lumen of the endoplasmic reticulum (ER) has resident proteins that are critical to perform the various tasks of the ER such as protein maturation and lipid metabolism. -

KDELR2 (NM 006854) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RG200007 KDELR2 (NM_006854) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: KDELR2 (NM_006854) Human Tagged ORF Clone Tag: TurboGFP Symbol: KDELR2 Synonyms: ELP-1; ELP1; ERD2.2; OI21 Vector: pCMV6-AC-GFP (PS100010) E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RG200007 representing NM_006854 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGAACATTTTCCGGCTGACTGGGGACCTGTCCCACCTGGCGGCCATCGTCATCCTGCTGCTGAAGATCT GGAAGACGCGCTCCTGCGCCGGTATTTCTGGGAAAAGCCAGCTTCTGTTTGCACTGGTCTTCACAACTCG TTACCTGGATCTTTTTACTTCATTTATTTCATTGTATAACACATCTATGAAGGTTATCTACCTTGCCTGC TCCTATGCCACAGTGTACCTGATCTACCTGAAATTTAAGGCAACCTACGATGGAAATCATGATACCTTCC GAGTGGAGTTTCTGGTGGTCCCTGTGGGAGGCCTCTCATTTTTAGTTAATCACGATTTCTCTCCTCTTGA GATCCTCTGGACCTTCTCCATCTACCTGGAGTCCGTGGCTATCCTTCCGCAGCTGTTTATGATCAGCAAG ACTGGGGAGGCCGAGACCATCACCACCCACTACCTGTTCTTCCTGGGCCTCTATCGTGCTTTGTATCTTG TCAACTGGATCTGGCGCTTCTACTTTGAGGGCTTCTTTGACCTCATTGCTGTGGTGGCCGGCGTAGTCCA GACCATCCTATACTGTGACTTCTTCTACTTGTACATTACAAAAGTACTCAAGGGAAAGAAGCTCAGTTTG CCAGCA ACGCGTACGCGGCCGCTCGAG - GFP Tag - GTTTAA Protein Sequence: >RG200007 representing NM_006854 Red=Cloning site Green=Tags(s) MNIFRLTGDLSHLAAIVILLLKIWKTRSCAGISGKSQLLFALVFTTRYLDLFTSFISLYNTSMKVIYLAC SYATVYLIYLKFKATYDGNHDTFRVEFLVVPVGGLSFLVNHDFSPLEILWTFSIYLESVAILPQLFMISK -

Examining Differences of Ubiquitination on KDEL Receptor Isoform Expression Zachary Dixon Mentored by Dr
Examining differences of ubiquitination on KDEL receptor isoform expression Zachary Dixon Mentored by Dr. Emily Wires and Dr. Brandon Harvey Introduction Materials and Methods (cont.) Results (cont.) The disruption of vital cellular processes, commonly supported by Following the identification of putative PTMs (Figure 2), KDELrs were probed Flag Pull-Down Flow-Through the transport of proteins emanant from the endoplasmic reticulum for ubiquitination. SY5Y cells were plated in a 24–well plate, and KDELr plasmids (ER), is a primary factor in disease pathogenesis. Key proteins in this were transfected into cells with no additional treatment, vehicle, or a proteasomal R1 R2 R1 R2 R1 R2 R1 R1 R1 R1 R2 R2 R2 R2 R1 transport are KDEL receptors (KDELrs). Post-translation, nascent inhibitor (MG132). Cells were then lysed and collected for immunoprecipitation R2 MANF proteins are folded by ER chaperones, a subgroup of ER-resident (IP). IP experiments were performed with magnetic beads incubated with FLAG- MANF proteins (ERPs), and moved them to the ER membrane (Trychta et antibodies, followed by PBS-T washes. Samples were centrifuged, incubated with al., 2018). They may also undergo post-translational modifications cell culture media, and eluted. Samples were then run on a 4–12% Bis-Tris gel and 51 kDa (PTMs). Post-secretion from the ER, they are trafficked through the proteins were transferred onto polyvinylidene difluoride (PVDF) membranes. For IgG Golgi, and to their final destination. However, ERPs are unique in this analysis of KDELr isoform relative abundance, PVDF membranes were probed 39 kDa regard; ERPs return from the Golgi to the ER upon interaction with a with IR secondary antibodies and scanned with an infrared scanner. -

KDELR2 Antibody (Aa81-130) Rabbit Polyclonal Antibody Catalog # ALS16666
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 KDELR2 Antibody (aa81-130) Rabbit Polyclonal Antibody Catalog # ALS16666 Specification KDELR2 Antibody (aa81-130) - Product Information Application IHC, IF, WB Primary Accession P33947 Other Accession 11014 Reactivity Human, Mouse, Rat Host Rabbit Clonality Polyclonal Isotype IgG Calculated MW 24422 Anti-KDELR2 antibody IHC staining of human brain, cortex. KDELR2 Antibody (aa81-130) - Additional Information Gene ID 11014 Other Names KDELR2, ERD2-like protein 1, ERD2.2, ERD-2-like protein, KDEL receptor 2, ELP-1 Immunofluorescence of A549 cells, using ERD22 Antibody. Target/Specificity ERD22 Antibody detects endogenous levels of total ERD22 protein. Reconstitution & Storage PBS (without Mg2+, Ca2+), pH 7.4, 150 mM sodium chloride, 0.02% sodium azide, 50% glycerol. Store at -20°C for up to one year. Precautions KDELR2 Antibody (aa81-130) is for research use only and not for use in diagnostic or therapeutic procedures. KDELR2 Antibody (aa81-130) - Protein Western blot of extracts from HUVEC/COLO Information cells, using ERD22 Antibody. Name KDELR2 KDELR2 Antibody (aa81-130) - Background Synonyms ERD2.2 {ECO:0000303|PubMed:1325562} Required for the retention of luminal endoplasmic reticulum proteins. Determines Function the specificity of the luminal ER protein Receptor for the C-terminal sequence motif retention system. Also required for normal Page 1/2 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 K-D-E-L that is present on endoplasmic vesicular traffic through the Golgi. This reticulum resident proteins and that receptor recognizes K-D-E-L. -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

CREB-Dependent Transcription in Astrocytes: Signalling Pathways, Gene Profiles and Neuroprotective Role in Brain Injury
CREB-dependent transcription in astrocytes: signalling pathways, gene profiles and neuroprotective role in brain injury. Tesis doctoral Luis Pardo Fernández Bellaterra, Septiembre 2015 Instituto de Neurociencias Departamento de Bioquímica i Biologia Molecular Unidad de Bioquímica y Biologia Molecular Facultad de Medicina CREB-dependent transcription in astrocytes: signalling pathways, gene profiles and neuroprotective role in brain injury. Memoria del trabajo experimental para optar al grado de doctor, correspondiente al Programa de Doctorado en Neurociencias del Instituto de Neurociencias de la Universidad Autónoma de Barcelona, llevado a cabo por Luis Pardo Fernández bajo la dirección de la Dra. Elena Galea Rodríguez de Velasco y la Dra. Roser Masgrau Juanola, en el Instituto de Neurociencias de la Universidad Autónoma de Barcelona. Doctorando Directoras de tesis Luis Pardo Fernández Dra. Elena Galea Dra. Roser Masgrau In memoriam María Dolores Álvarez Durán Abuela, eres la culpable de que haya decidido recorrer el camino de la ciencia. Que estas líneas ayuden a conservar tu recuerdo. A mis padres y hermanos, A Meri INDEX I Summary 1 II Introduction 3 1 Astrocytes: physiology and pathology 5 1.1 Anatomical organization 6 1.2 Origins and heterogeneity 6 1.3 Astrocyte functions 8 1.3.1 Developmental functions 8 1.3.2 Neurovascular functions 9 1.3.3 Metabolic support 11 1.3.4 Homeostatic functions 13 1.3.5 Antioxidant functions 15 1.3.6 Signalling functions 15 1.4 Astrocytes in brain pathology 20 1.5 Reactive astrogliosis 22 2 The transcription -
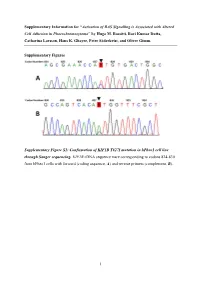
Activation of RAS Signalling Is Associated with Altered Cell Adhesion in Phaeochromocytoma” by Hugo M
Supplementary Information for “Activation of RAS Signalling is Associated with Altered Cell Adhesion in Phaeochromocytoma” by Hugo M. Rossitti, Ravi Kumar Dutta, Catharina Larsson, Hans K. Ghayee, Peter Söderkvist, and Oliver Gimm. Supplementary Figures Supplementary Figure S1: Confirmation of KIF1B T827I mutation in hPheo1 cell line through Sanger sequencing. KIF1B cDNA sequence trace corresponding to codons 824-830 from hPheo1 cells with forward (coding sequence, A) and reverse primers (complement, B). 1 Supplementary Figure S2: Confirmation of NRAS Q61K mutation in hPheo1 cell line through Sanger sequencing. NRAS cDNA sequence trace corresponding to codons 56-67 from hPheo1 cells with forward (coding sequence, A) and reverse primers (complement, B). 2 Supplementary Figure S3: CCND1 gene expression and hPheo1 proliferation. A: Expression of CCND1 mRNA assessed by RT-qPCR and presented as fold change (2-ΔΔCT, mean ± standard error of the mean). B: Cell counts at 1, 2, and 3 days after plating (corresponding to 72, 96 and 120 hours posttransfection, respectively) of control- or siNRAS#1-transfected hPheo1 cells expressed as fold change of the number of cells plated at day 0 (48 hours posttransfection; mean ± standard deviation). All results are from three independent siRNA experiments. 3 Supplementary Tables Supplementary Table S1: List of transcript cluster IDs significantly upregulated in hPheo1 by siNRAS treatment (comparison: siNRAS versus control-transfected hPheo1; ANOVA p < 0.05, FDR < 0.25, fold change < -1.5 or > 1.5). Transcript -

The Function of KDEL Receptors As UPR Genes in Disease
International Journal of Molecular Sciences Review The Function of KDEL Receptors as UPR Genes in Disease Emily S. Wires *,†, Kathleen A. Trychta †, Lacey M. Kennedy † and Brandon K. Harvey *,† Molecular Mechanisms of Cellular Stress and Inflammation, Intramural Research Program, National Institute on Drug Abuse, Baltimore, MD 21224, USA; [email protected] (K.A.T.); [email protected] (L.M.K.) * Correspondence: [email protected] (E.S.W.); [email protected] (B.K.H.) † These authors contributed equally. Abstract: The KDEL receptor retrieval pathway is essential for maintaining resident proteins in the endoplasmic reticulum (ER) lumen. ER resident proteins serve a variety of functions, including protein folding and maturation. Perturbations to the lumenal ER microenvironment, such as calcium depletion, can cause protein misfolding and activation of the unfolded protein response (UPR). Additionally, ER resident proteins are secreted from the cell by overwhelming the KDEL receptor retrieval pathway. Recent data show that KDEL receptors are also activated during the UPR through the IRE1/XBP1 signaling pathway as an adaptive response to cellular stress set forth to reduce the loss of ER resident proteins. This review will discuss the emerging connection between UPR activation and KDEL receptors as it pertains to ER proteostasis and disease states. Keywords: KDEL receptor; endoplasmic reticulum; unfolded protein response; ER resident proteins; disease; exodosis 1. Introduction Citation: Wires, E.S.; Trychta, K.A.; Kennedy, L.M.; Harvey, B.K. The The endoplasmic reticulum (ER) is a dynamic intracellular organelle integral to cel- Function of KDEL Receptors as UPR lular homeostasis. Although the ER plays critical roles in lipid synthesis [1], calcium Genes in Disease. -

Case of 7P22. 1 Microduplication Detected by Whole Genome Microarray (REVEAL) in Workup of Child Diagnosed with Autism
Hindawi Publishing Corporation Case Reports in Genetics Volume 2015, Article ID 212436, 6 pages http://dx.doi.org/10.1155/2015/212436 Case Report Case of 7p22.1 Microduplication Detected by Whole Genome Microarray (REVEAL) in Workup of Child Diagnosed with Autism Veronica Goitia,1 Marcial Oquendo,1 and Robert Stratton2 1 Department of Pediatrics, Driscoll Children’s Hospital, Corpus Christi, TX 78411, USA 2Department of Medical Genetics, Driscoll Children’s Hospital, Corpus Christi, TX 78411, USA Correspondence should be addressed to Veronica Goitia; [email protected] Received 2 October 2014; Revised 1 February 2015; Accepted 6 March 2015 Academic Editor: Mohnish Suri Copyright © 2015 Veronica Goitia et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Introduction. More than 60 cases of 7p22 duplications and deletions have been reported with over 16 of them occurring without concomitant chromosomal abnormalities. Patient and Methods. We report a 29-month-old male diagnosed with autism. Whole genome chromosome SNP microarray (REVEAL) demonstrated a 1.3 Mb interstitial duplication of 7p22.1 ->p22.1 arr 7p22.1 (5,436,367–6,762,394), the second smallest interstitial 7p duplication reported to date. This interval included 14 OMIM annotated genes (FBXL18, ACTB, FSCN1, RNF216, OCM, EIF2AK1, AIMP2, PMS2, CYTH3, RAC1, DAGLB, KDELR2, GRID2IP, and ZNF12). Results. Our patient presented features similar to previously reported cases with 7p22 duplication, including brachycephaly, prominent ears, cryptorchidism, speech delay, poor eye contact, and outburst of aggressive behavior with autism-like features. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Anti-KDEL Receptor 2 Antibody (ARG66369)
Product datasheet [email protected] ARG66369 Package: 100 μg anti-KDEL Receptor 2 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes KDEL Receptor 2 Tested Reactivity Hu, Ms, Rat Tested Application ICC/IF, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name KDEL Receptor 2 Antigen Species Human Immunogen Synthetic peptide corresponding to aa. 50-130 of Human KDEL Receptor 2. Conjugation Un-conjugated Alternate Names ERD2-like protein 1; ELP-1; KDEL endoplasmic reticulum protein retention receptor 2; KDEL receptor 2; ERD2.2; ER lumen protein-retaining receptor 2 Application Instructions Application table Application Dilution ICC/IF 1:200 - 1:1000 WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 24 kDa Observed Size 24 kDa Properties Form Liquid Purification Affinity purification with immunogen. Buffer PBS, 0.02% Sodium azide, 50% Glycerol and 0.5% BSA. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol and 0.5% BSA Concentration 1 mg/ml Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. www.arigobio.com 1/2 Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol KDELR2 Gene Full Name KDEL (Lys-Asp-Glu-Leu) endoplasmic reticulum protein retention receptor 2 Background Retention of resident soluble proteins in the lumen of the endoplasmic reticulum (ER) is achieved in both yeast and animal cells by their continual retrieval from the cis-Golgi, or a pre-Golgi compartment.