Huey 2008 02Thesis.Pdf (9.880Mb)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Queensland Public Boat Ramps
Queensland public boat ramps Ramp Location Ramp Location Atherton shire Brisbane city (cont.) Tinaroo (Church Street) Tinaroo Falls Dam Shorncliffe (Jetty Street) Cabbage Tree Creek Boat Harbour—north bank Balonne shire Shorncliffe (Sinbad Street) Cabbage Tree Creek Boat Harbour—north bank St George (Bowen Street) Jack Taylor Weir Shorncliffe (Yundah Street) Cabbage Tree Creek Boat Harbour—north bank Banana shire Wynnum (Glenora Street) Wynnum Creek—north bank Baralaba Weir Dawson River Broadsound shire Callide Dam Biloela—Calvale Road (lower ramp) Carmilla Beach (Carmilla Creek Road) Carmilla Creek—south bank, mouth of creek Callide Dam Biloela—Calvale Road (upper ramp) Clairview Beach (Colonial Drive) Clairview Beach Moura Dawson River—8 km west of Moura St Lawrence (Howards Road– Waverley Creek) Bund Creek—north bank Lake Victoria Callide Creek Bundaberg city Theodore Dawson River Bundaberg (Kirby’s Wall) Burnett River—south bank (5 km east of Bundaberg) Beaudesert shire Bundaberg (Queen Street) Burnett River—north bank (downstream) Logan River (Henderson Street– Henderson Reserve) Logan Reserve Bundaberg (Queen Street) Burnett River—north bank (upstream) Biggenden shire Burdekin shire Paradise Dam–Main Dam 500 m upstream from visitors centre Barramundi Creek (Morris Creek Road) via Hodel Road Boonah shire Cromarty Creek (Boat Ramp Road) via Giru (off the Haughton River) Groper Creek settlement Maroon Dam HG Slatter Park (Hinkson Esplanade) downstream from jetty Moogerah Dam AG Muller Park Groper Creek settlement Bowen shire (Hinkson -

§4-71-6.5 LIST of CONDITIONALLY APPROVED ANIMALS November
§4-71-6.5 LIST OF CONDITIONALLY APPROVED ANIMALS November 28, 2006 SCIENTIFIC NAME COMMON NAME INVERTEBRATES PHYLUM Annelida CLASS Oligochaeta ORDER Plesiopora FAMILY Tubificidae Tubifex (all species in genus) worm, tubifex PHYLUM Arthropoda CLASS Crustacea ORDER Anostraca FAMILY Artemiidae Artemia (all species in genus) shrimp, brine ORDER Cladocera FAMILY Daphnidae Daphnia (all species in genus) flea, water ORDER Decapoda FAMILY Atelecyclidae Erimacrus isenbeckii crab, horsehair FAMILY Cancridae Cancer antennarius crab, California rock Cancer anthonyi crab, yellowstone Cancer borealis crab, Jonah Cancer magister crab, dungeness Cancer productus crab, rock (red) FAMILY Geryonidae Geryon affinis crab, golden FAMILY Lithodidae Paralithodes camtschatica crab, Alaskan king FAMILY Majidae Chionocetes bairdi crab, snow Chionocetes opilio crab, snow 1 CONDITIONAL ANIMAL LIST §4-71-6.5 SCIENTIFIC NAME COMMON NAME Chionocetes tanneri crab, snow FAMILY Nephropidae Homarus (all species in genus) lobster, true FAMILY Palaemonidae Macrobrachium lar shrimp, freshwater Macrobrachium rosenbergi prawn, giant long-legged FAMILY Palinuridae Jasus (all species in genus) crayfish, saltwater; lobster Panulirus argus lobster, Atlantic spiny Panulirus longipes femoristriga crayfish, saltwater Panulirus pencillatus lobster, spiny FAMILY Portunidae Callinectes sapidus crab, blue Scylla serrata crab, Samoan; serrate, swimming FAMILY Raninidae Ranina ranina crab, spanner; red frog, Hawaiian CLASS Insecta ORDER Coleoptera FAMILY Tenebrionidae Tenebrio molitor mealworm, -

NW Queensland Water Supply Strategy Investigation
NW Queensland Water Supply Strategy Investigation Final Consultant Report 9 March 2016 Document history Author/s Romy Greiner Brett Twycross Rohan Lucas Checked Adam Neilly Approved Brett Twycross Contact: Name Alluvium Consulting Australia ABN 76 151 119 792 Contact person Brett Twycross Ph. (07) 4724 2170 Email [email protected] Address 412 Flinders Street Townsville QLD 4810 Postal address PO Box 1581 Townsville QLD 4810 Ref Contents 1 Introduction 1 2 Methodology 2 2.1 Geographic scope and relevant regional characteristics 2 2.2 Situation and vulnerability analysis 3 2.3 Multi criteria decision analysis 5 2.3.1 The principles of multi criteria decision making 5 2.3.2 Quantitative criteria 7 2.3.3 Qualitative criteria 8 3 Situation analysis: Water demand and supply 12 3.1 Overview 12 3.2 Urban water demand and supply 14 3.2.1 Mount Isa 14 3.2.2 Cloncurry 15 3.3 Mining and mineral processing water demand and supply 16 3.3.1 Mount Isa precinct 16 3.3.2 Cloncurry precinct 17 3.4 Agriculture 18 3.5 Uncommitted water 19 3.6 Projected demand and water security 19 3.7 Vulnerability to water shortages 20 4 Water infrastructure alternatives 21 4.1 New water storage in the upper Cloncurry River catchment 23 4.1.1 Cave Hill Dam 23 4.1.2 Black Fort Dam 25 4.1.3 Painted Rock Dam 26 4.1.4 Slaty Creek 27 4.1.5 Combination of Black Fort Dam and Slaty Creek 27 4.2 Increasing the capacity of the Lake Julius water supply 28 4.3 Utilising currently unused water storage infrastructure 30 4.3.1 Corella Dam 30 4.3.2 Lake Mary Kathleen 31 5 Ranking -

Fishes of the King Edward River in the Kimberley Region, Western Australia
Records of the Western Australian Museum 25: 351–368 (2010). Fishes of the King Edward River in the Kimberley region, Western Australia David L. Morgan Freshwater Fish Group, Centre for Fish and Fisheries Research, Murdoch University, Murdoch, Western Australia 6150, Australia. E-mail: [email protected] Abstract – The King Edward River, in the far north of the Kimberley region of Western Australia drains approximately 10,000 km2 and discharges into the Timor Sea near the town of Kalumburu. This study represents an ichthyological survey of the river’s freshwaters and revealed that the number of freshwater fishes of the King Edward River is higher than has previously been recorded for a Western Australian river. Twenty-six strictly freshwater fish species were recorded, which is three species higher than the much larger Fitzroy River in the southern Kimberley. The study also identified a number of range extensions, including Butler’s Grunter and Shovel-nosed Catfish to the west, and the Slender Gudgeon to the north and east. A possibly undescribed species of glassfish, that differs morphologically from described species in arrangement of head spines, fin rays, as well as relative body measurements, is reported. A considerable proportion of Jenkins’ Grunter, which is widespread throughout the system but essentially restricted to main channel sites, had ‘blubber-lips’. There were significant differences in the prevailing fish fauna of the different reaches of the King Edward River system. Thus fish associations in the upper King Edward River main channel were significantly different to those in the tributaries and the main channel of the Carson River. -

Lakes Argyle and Kununurra Wetlands Ramsar Site Ecological Character Description
Lakes Argyle and Kununurra Ramsar Site Ecological Character Description Citation: Hale, J. and Morgan, D., 2010, Ecological Character Description for the Lakes Argyle and Kununurra Ramsar Site. Report to the Department of Sustainability, Environment, Water, Population and Communities, Canberra. Acknowledgements: Danny Rogers, Australasian Waders Studies Group (expert advice) Halina Kobryn, Murdoch University (mapping and GIS) The steering committee was comprised of representatives of the following organisations: • Department of the Environment, Water, Heritage and the Arts • WA Department of Environment and Conservation (Kununurra) • WA Department of Water (Kununurra) • Shire of Wyndham East Kimberley Introductory Notes This Ecological Character Description (ECD Publication) has been prepared in accordance with the National Framework and Guidance for Describing the Ecological Character of Australia’s Ramsar Wetlands (National Framework) (Department of the Environment, Water, Heritage and the Arts, 2008). The Environment Protection and Biodiversity Conservation Act 1999 (EPBC Act) prohibits actions that are likely to have a significant impact on the ecological character of a Ramsar wetland unless the Commonwealth Environment Minister has approved the taking of the action, or some other provision in the EPBC Act allows the action to be taken. The information in this ECD Publication does not indicate any commitment to a particular course of action, policy position or decision. Further, it does not provide assessment of any particular action within the meaning of the Environment Protection and Biodiversity Conservation Act 1999 (Cth), nor replace the role of the Minister or his delegate in making an informed decision to approve an action. The Water Act 2007 requires that in preparing the [Murray-Darling] Basin Plan, the Murray Darling Basin Authority (MDBA) must take into account Ecological Character Descriptions of declared Ramsar wetlands prepared in accordance with the National Framework. -

Strategic Framework December 2019 CS9570 12/19
Department of Natural Resources, Mines and Energy Queensland bulk water opportunities statement Part A – Strategic framework December 2019 CS9570 12/19 Front cover image: Chinaman Creek Dam Back cover image: Copperlode Falls Dam © State of Queensland, 2019 The Queensland Government supports and encourages the dissemination and exchange of its information. The copyright in this publication is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. Under this licence you are free, without having to seek our permission, to use this publication in accordance with the licence terms. You must keep intact the copyright notice and attribute the State of Queensland as the source of the publication. For more information on this licence, visit https://creativecommons.org/licenses/by/4.0/. The information contained herein is subject to change without notice. The Queensland Government shall not be liable for technical or other errors or omissions contained herein. The reader/user accepts all risks and responsibility for losses, damages, costs and other consequences resulting directly or indirectly from using this information. Hinze Dam Queensland bulk water opportunities statement Contents Figures, insets and tables .....................................................................iv 1. Introduction .............................................................................1 1.1 Purpose 1 1.2 Context 1 1.3 Current scope 2 1.4 Objectives and principles 3 1.5 Objectives 3 1.6 Principles guiding Queensland Government investment 5 1.7 Summary of initiatives 9 2. Background and current considerations ....................................................11 2.1 History of bulk water in Queensland 11 2.2 Current policy environment 12 2.3 Planning complexity 13 2.4 Drivers of bulk water use 13 3. -

Queensland Government Gazette
Queensland Government Gazette PP 451207100087 PUBLISHED BY AUTHORITY ISSN 0155-9370 Vol. CCCXXXIV] FRIDAY, 21 NOVEMBER, 2003 EDEN RITCHIE RECRUITMENT - focused on Accounting, Executive and IT Recruitment Give us a bell if you need people over Christmas... (or any other time) www.edenritchie.com.au H&J 8329 phone: (07) 3236 0033 fax: (07) 3236 0099 email: info@ edenritchie.com.au [895] Queensland Government Gazette PP 451207100087 PUBLISHED BY AUTHORITY ISSN 0155-9370 Vol. CCCXXXIV] FRIDAY, 21 NOVEMBER, 2003 [No. 56 Local Government Act 1993 NEBO SHIRE COUNCIL (MAKING OF LOCAL LAW) NOTICE (NO. 2) 2003 Title 1. This notice may be cited as the Nebo Shire Council (Making of Local Law) Notice (No. 2) 2003. Commencement © The State of Queensland 2003. 2. This notice commences on the date it is published in the Copyright protects this publication. Except for purposes permitted by the Gazette. Copyright Act, reproduction by whatever means is prohibited without the prior written permission of Goprint. Making of local law Inquiries should be addressed to Goprint, Publications and Retail, 371 3. Pursuant to the provisions of the Local Government Act 1993 Vulture Street, Woolloongabba, 4102. the Nebo Shire Council made Keeping and Control of Animals (Amendment) Local Law (No.1) 2003 by resolution on 23 October, 2003 which amends Local Law No.12 (Keeping and Control of Animals). —————— BRISBANE Inspection Printed and Published by Government Printer, 4. A certified copy of the local law is open to inspection at the Vulture Street, Woolloongabba local government’s public office and at the Department’s 21 November, 2003 State office. -

Southern Gulf, Queensland
Biodiversity Summary for NRM Regions Species List What is the summary for and where does it come from? This list has been produced by the Department of Sustainability, Environment, Water, Population and Communities (SEWPC) for the Natural Resource Management Spatial Information System. The list was produced using the AustralianAustralian Natural Natural Heritage Heritage Assessment Assessment Tool Tool (ANHAT), which analyses data from a range of plant and animal surveys and collections from across Australia to automatically generate a report for each NRM region. Data sources (Appendix 2) include national and state herbaria, museums, state governments, CSIRO, Birds Australia and a range of surveys conducted by or for DEWHA. For each family of plant and animal covered by ANHAT (Appendix 1), this document gives the number of species in the country and how many of them are found in the region. It also identifies species listed as Vulnerable, Critically Endangered, Endangered or Conservation Dependent under the EPBC Act. A biodiversity summary for this region is also available. For more information please see: www.environment.gov.au/heritage/anhat/index.html Limitations • ANHAT currently contains information on the distribution of over 30,000 Australian taxa. This includes all mammals, birds, reptiles, frogs and fish, 137 families of vascular plants (over 15,000 species) and a range of invertebrate groups. Groups notnot yet yet covered covered in inANHAT ANHAT are notnot included included in in the the list. list. • The data used come from authoritative sources, but they are not perfect. All species names have been confirmed as valid species names, but it is not possible to confirm all species locations. -

Freshwater Fishes of Kakadu National Park and the Impact of Sea Level Rise
Freshwater fishes of Kakadu National Park and the impact of sea level rise What is the issue? (both brackish and freshwater) may persist in the landscape for less time. While some estuarine species Sixty two species of freshwater fish have been may potentially benefit from expanded tidal habitat, recorded from Kakadu National Park. For 11 of some lowland freshwater species, particularly those these species, between 15% and 40% of their total largely confined to this habitat type, would appear to predicted distribution occurs within the Park. Many be vulnerable. This study, funded under the Northern freshwater fish species in the Park are likely to be Australia Hub of the Australian Government’s impacted by climate change, especially saltwater National Environmental Research Program, examined intrusion onto freshwater floodplains as a result of the direct effects of sea level rise on floodplain sea level rise and increased storm surge. freshwater fish species. Sea level rise is predicted to cause rapid and substantial habitat changes. In particular, affected What did the research do? freshwater wetlands are predicted to change to To predict the extent of impact of sea level rise open saline mudflats. Floodplain and mudflat on floodplain fishes the team developed a model habitats may also experience higher temperatures incorporating topographic information for the and faster evaporation rates and seasonal wetlands lowland rivers, floodplains and estuaries of the Park and the predicted distribution data for 55 freshwater fish species. While climate change scientists are confident that sea levels will rise, due to the complexity of climate systems, the exact rate and amount of change is unknown. -
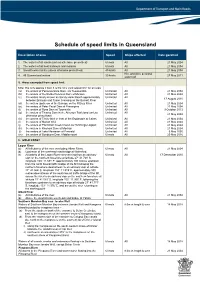
Schedule of Speed Limits in Queensland
Schedule of speed limits in Queensland Description of area Speed Ships affected Date gazetted 1. The waters of all canals (unless otherwise prescribed) 6 knots All 21 May 2004 2. The waters of all boat harbours and marinas 6 knots All 21 May 2004 3. Smooth water limits (unless otherwise prescribed) 40 knots All 21 May 2004 Hire and drive personal 4. All Queensland waters 30 knots 27 May 2011 watercraft 5. Areas exempted from speed limit Note: this only applies if item 3 is the only valid speed limit for an area (a) the waters of Perserverance Dam, via Toowoomba Unlimited All 21 May 2004 (b) the waters of the Bjelke Peterson Dam at Murgon Unlimited All 21 May 2004 (c) the waters locally known as Sandy Hook Reach approximately Unlimited All 17 August 2010 between Branyan and Tyson Crossing on the Burnett River (d) the waters upstream of the Barrage on the Fitzroy River Unlimited All 21 May 2004 (e) the waters of Peter Faust Dam at Proserpine Unlimited All 21 May 2004 (f) the waters of Ross Dam at Townsville Unlimited All 9 October 2013 (g) the waters of Tinaroo Dam in the Atherton Tableland (unless Unlimited All 21 May 2004 otherwise prescribed) (h) the waters of Trinity Inlet in front of the Esplanade at Cairns Unlimited All 21 May 2004 (i) the waters of Marian Weir Unlimited All 21 May 2004 (j) the waters of Plantation Creek known as Hutchings Lagoon Unlimited All 21 May 2004 (k) the waters in Kinchant Dam at Mackay Unlimited All 21 May 2004 (l) the waters of Lake Maraboon at Emerald Unlimited All 6 May 2005 (m) the waters of Bundoora Dam, Middlemount 6 knots All 20 May 2016 6. -

Fishes of the King Edward and Carson Rivers with Their Belaa and Ngarinyin Names
Fishes of the King Edward and Carson Rivers with their Belaa and Ngarinyin names By David Morgan, Dolores Cheinmora Agnes Charles, Pansy Nulgit & Kimberley Language Resource Centre Freshwater Fish Group CENTRE FOR FISH & FISHERIES RESEARCH Kimberley Language Resource Centre Milyengki Carson Pool Dolores Cheinmora: Nyarrinjali, kaawi-lawu yarn’ nyerreingkana, Milyengki-ngûndalu. Waj’ nyerreingkana, kaawi-ku, kawii amûrike omûrung, yilarra a-mûrike omûrung. Agnes Charles: We are here at Milyengki looking for fish. He got one barramundi, a small one. Yilarra is the barramundi’s name. Dolores Cheinmora: Wardi-di kala’ angbûnkû naa? Agnes Charles: Can you see the fish, what sort of fish is that? Dolores Cheinmora: Anja kûkûridingei, Kalamburru-ngûndalu. Agnes Charles: This fish, the Barred Grunter, lives in the Kalumburu area. Title: Fishes of the King Edward and Carson Rivers with their Belaa and Ngarinyin names Authors: D. Morgan1 D. Cheinmora2, A. Charles2, Pansy Nulgit3 & Kimberley Language Resource Centre4 1Centre for Fish & Fisheries Research, Murdoch University, South St Murdoch WA 6150 2Kalumburu Aboriginal Corporation 3Kupungari Aboriginal Corporation 4Siobhan Casson, Margaret Sefton, Patsy Bedford, June Oscar, Vicki Butters - Kimberley Language Resource Centre, Halls Creek, PMB 11, Halls Creek WA 6770 Project funded by: Land & Water Australia Photographs on front cover: Lower King Edward River Long-nose Grunter (inset). July 2006 Land & Water Australia Project No. UMU22 Fishes of the King Edward River - Centre for Fish & Fisheries Research, Murdoch University / Kimberley Language Resource Centre 2 Acknowledgements Most importantly we would like to thank the people of the Kimberley, particularly the Traditional Owners at Kalumburu and Prap Prap. This project would not have been possible without the financial support of Land & Water Australia. -

Mount Isa Regional Water Supply Security Assessment CS9703 12/19
Department of Natural Resources, Mines and Energy Mount Isa regional water supply security assessment CS9703 12/19 This publication has been compiled by the Department of Natural Resources, Mines and Energy © State of Queensland, 2019. The Queensland Government supports and encourages the dissemination and exchange of its information. The copyright in this publication is licensed under a Creative Commons Attribution 4.0 Australia (CC BY 4.0) licence. Under this licence you are free, without having to seek our permission, to use this publication in accordance with the licence terms. You must keep intact the copyright notice and attribute the State of Queensland as the source of the publication. Note: Some content in this publication may have different licence terms as indicated. For more information on this licence, visit http://creativecommons.org/licenses/by/4.0 The information contained herein is subject to change without notice. The Queensland Government shall not be liable for technical or other errors or omissions contained herein. The reader/user accepts all risks and responsibilities for losses, damages, costs and other consequences resulting directly or indirectly from using this information. The Queensland Government is committed to providing accessible services to Queenslanders from all culturally and linguistically diverse backgrounds. If you have difficulty in understanding this document, you can contact us within Australia on 13QGOV (13 74 68) and we will arrange an interpreter to effectively communicate the report to you. Image courtesy of Tourism and Events Queensland Introduction Mount Isa is a mining and commercial service centre in the heart of the Carpentaria Mount Isa Minerals Province in north-west Queensland.