Arxiv:1701.02495V2 [Cond-Mat.Mtrl-Sci] 6 Jul 2017
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MBN Explorer Users' Guide
Ilia A. Solov’yov, Gennady Sushko and Andrey V. Solov’yov MBN Explorer Users’ Guide Version 3.0 Ilia A. Solov'yov Department of Physics, Chemistry and Pharmacy University of Southern Denamrk Odense, Denmark Gennady Sushko MBN Research Center gGmbH Frankfurt, Germany Andrey V. Solov'yov MBN Research Center gGmbH Frankfurt, Germany MesoBioNano Science Publishing ⃝c MBN Research Center gGmbH 2017. All rights reserved. This book is subject to a copyright agreement, where the Publisher reserves the rights of translation, copying, reprinting, advertising and reproduction of the whole book or its parts by physical, electronic or similar methodology developed by today or hereafter. The use of specic and general terms, abbreviates, descriptive and registered names, trade and service marks, included in this book does not imply that such names are exempt from the relevant protective laws and regulations and therefore free for general use. The responsibility to inquire about such possibility is with the individual customer. The information provided in this book is true and accurate to the best knowledge of the publisher and the authors at the date of the publication. The publisher and the authors do not provide any warranty with respect to the material contained in this book or for any error or omissions that may have been made. The publisher and the authors do not take responsibility for any damages related to the use of the material contained in this book. The publisher remains neutral with regard to any jurisdictional claims in published maps and institutional aliations. The registered company is MBN Research Center gGmbH. The company address is Altenhöferallee 3, 60438 Frankfurt am Main, Germany. -

Modeling the Effect of Ion-Induced Shock Waves and DNA Breakage
Modeling the effect of ion-induced shock waves and DNA breakage with the reactive CHARMM force field Ida Friis,1 Alexey V. Verkhovtsev,2, ∗ Ilia A. Solov’yov,3, ∗ and Andrey V. Solov’yov2, ∗ 1Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark 2MBN Research Center, Altenh¨oferallee 3, 60438 Frankfurt am Main, Germany 3Department of Physics, Carl von Ossietzky Universit¨at Oldenburg, Carl-von-Ossietzky-Str. 9-11, 26129 Oldenburg, Germany Ion-induced DNA damage is an important effect underlying ion beam cancer therapy. This paper introduces the methodology of modeling DNA damage induced by a shock wave caused by a projectile ion. Specifically it is demonstrated how single- and double strand breaks in a DNA molecule could be described by the reactive CHARMM (rCHARMM) force field implemented in the program MBN Explorer. The entire work flow of performing the shock wave simulations, including obtaining the crucial simulation parameters, is described in seven steps. Two exemplary analyses are provided for a case study simulation serving to: (i) quantify the shock wave propagation, and (ii) describe the dynamics of formation of DNA breaks. The paper concludes by discussing the computational cost of the simulations and revealing the possible maximal computational time for different simulation setups. I. INTRODUCTION changes of their topology and molecular composition [9, 11, 12]. It takes into account additional parameters Classical molecular dynamics (MD) enables studying of the system, such as bond dissociation energy, bond of structure and dynamics of biological systems and inor- order and the valence of atoms. -

Research Report Hochleistungsrechner in Hessen 2010/2011 Inhalt
Research Report Hochleistungsrechner in Hessen 2010/2011 Inhalt Vorwort zum Jahresbericht 2010-2011 ......................................................................................................................................................................................... 7 Die Hochleistungsrechner Der Hessische Hochleistungsrechner an der Technischen Universität Darmstadt ...................................................................................................8 Die Hochleistungsrechner (HLR) am Center for Scientific Computing (CSC) der Goethe-Universität ..................................................10 Anpassung des LINPACK-Benchmarks für den LOEWE-CSC ............................................................................................................................................12 Das Linux-Cluster im IT-Servicezentrum der Universität Kassel ........................................................................................................................................13 Die Hochleistungsrechner der GSI ........................................................................................................................................................................................................14 Die Rechencluster der Philipps-Universität Marburg.................................................................................................................................................................16 Das zentrale Hochleistungsrechen-Cluster der Justus-Liebig-Universität Gießen -
Advanced Materials Modelling and Characterization.Pdf
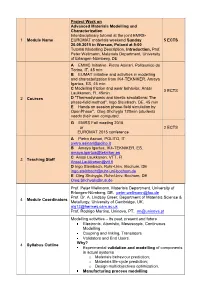
Project Work on Advanced Materials Modelling and Characterization Interdisciplinary tutorial at the joint EMRS- 1 Module Name EUROMAT materials weekend Sunday 5 ECTS 20.09.2015 in Warsaw, Poland at 9:00 Tutorial Modelling Description, Introduction, Prof. Peter Wellmann, Materials Department, University of Erlangen-Nürnberg, DE A EMMC Initiative, Pietro Asinari, Politecnico do Torino, IT, 45 min B EUMAT Initiative and activities in modelling and characterization from IK4-TEKNIKER, Amaya Igartua, ES, 45 min C Modelling friction and wear behavior, Anssi 3 ECTS Laukkanen, FI, 45min 2 Courses D "Thermodynamic and kinetic simulations: The phase-field method", Ingo Steinbach, DE, 45 min E Hands on session phase-field simulation by OpenPhase", Oleg Shchyglo 120min (students needs their own computer). G EMRS Fall meeting 2015 or 2 ECTS EUROMAT 2015 conference A Pietro Asinari, POLITO, IT [email protected] B Amaya Igartua, IK4-TEKNIKER, ES, [email protected] C Anssi Laukkanen, VTT, FI 3 Teaching Staff [email protected] D Ingo Steinbach, Ruhr-Univ. Bochum, DE [email protected] E Oleg Shchyglo, Ruhr-Univ. Bochum, DE [email protected] Prof. Peter Wellmann, Materials Department, University of Erlangen-Nürnberg, DE, [email protected] Prof. Dr. A. Lindsay Greer, Department of Materials Science & 4 Module Coordinators Metallurgy, University of Cambridge, UK, [email protected] Prof. Rodrigo Martins, Uninova, PT, [email protected] Modelling activities – its past, present and future Electronic, Atomistic, Mesoscopic, Continuous Modelling Coupling and linking, Translators Validators and End Users. Why? 4 Syllabus Outline Experimental validation and modelling of components in actual systems o Materials behaviour prediction; o Materials life-cycle prediction; o Design multiobjectives optimization. -

MBN Explorer Users' Guide Describes How to Run and to Use the Various Features of the Multiscale Program MBN Explorer
A universal program for multiscale simulation of complex molecular structure and dynamics MBN Explorer Users’ Guide Version 3.0 (March 31, 2017) Current editors: Ilia A. Solov’yov, Gennady Sushko and Andrey V. Solov’yov URL: www.mbnexplorer.com E-mail: [email protected] Description The MBN Explorer Users' Guide describes how to run and to use the various features of the multiscale program MBN Explorer. This guide includes the descrip- tion of the main features of the program, the manual how to use the program, the specication of input les and formats, and instructions on how to run the program on Windows, Linux/Unix and Macintosh platforms. Contents 1 Introduction 1 1.1 Features of MBN Explorer ..................... 1 1.2 MBN Explorer functionality .................... 3 1.3 Historical remarks ............................ 5 1.4 Terms and conditions .......................... 9 1.5 Third party terms and conditions ................... 18 1.5.1 VMD molle plugins ...................... 18 1.5.2 Mersenne twister ........................ 19 1.5.3 Kiss FFT ............................ 19 2 Getting Started 21 2.1 What is needed ............................. 21 2.2 MBN Explorer task le ....................... 21 2.2.1 Task le syntax ......................... 22 2.2.2 Required parameters ...................... 22 2.3 MBN Explorer task parameters . 22 2.3.1 General parameters ....................... 22 2.3.2 Input les ............................ 24 2.3.3 Output les and related parameters . 25 2.3.4 General numerical methods for interatomic interactions . 30 2.3.5 Ewald summation method for Coulomb interactions . 33 2.3.6 Fast particle mesh Ewald algorithm . 34 2.3.7 General molecular dynamics simulation . 36 2.3.8 Temperature control .....................