Commensal Microbiota Modulate Gene Expression in the Skin Jacquelyn S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Critical Evaluation of Gene Expression Changes in Human Tissues In
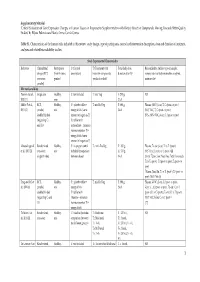
Supplementary Material ‘Critical Evaluation of Gene Expression Changes in Human Tissues in Response to Supplementation with Dietary Bioactive Compounds: Moving Towards Better-Quality Studies’ by Biljana Pokimica and María-Teresa García-Conesa Table S1. Characteristics of the human trials included in this review: study design, type of participants, control and intervention description, dose and duration of treatment, analyses and related bioavailability studies. Study Experimental Characteristics Reference Clinical trial Participants C (Control T (Treatment with Total daily dose, Bioavailability studies: type of sample, design (RCT, (health status, description) bioactive compounds, duration (d or h)1 compounds and (or) metabolites analysed, crossover, gender) products or diet) main results2 parallel) Mix meals and diets Persson I et al., Single arm Healthy, C: not included T: mix Veg T: 250 g, NR 2000 [1] men 21 d Møller P et al., RCT, Healthy, C1: placebo tablet + T: mix FruVeg T: 600 g, Plasma: (NS↑) β-car, T, C2 (post- vs pre-) 2003 [2] parallel, mix energy drink (same 24 d (NC) VitC, T, C2 (post- vs pre-) double blinded amount of sugars as T) (NS↓, 69%) VitC, β-car, C1 (post- vs pre-) (regarding C1 C2: tablet with and C2) antioxidants + minerals (same amount as T) + energy drink (same amount of sugars as T) Almendingen K Randomized, Healthy, C: no proper control T1,2: mix FruVeg T1: 300 g, Plasma: ↑α-car, β-car, T2 vs T1 (post-) et al., 2005 [3] crossover, mix included (comparison T2: 750 g, (NS↑) Lyc, Lut, T2 vs T1 (post-) [4] single -

Function in Vertebrates Receptor-Containing Adaptor
Evidence for Evolving Toll-IL-1 Receptor-Containing Adaptor Molecule Function in Vertebrates This information is current as Con Sullivan, John H. Postlethwait, Christopher R. Lage, of September 30, 2021. Paul J. Millard and Carol H. Kim J Immunol 2007; 178:4517-4527; ; doi: 10.4049/jimmunol.178.7.4517 http://www.jimmunol.org/content/178/7/4517 Downloaded from References This article cites 60 articles, 30 of which you can access for free at: http://www.jimmunol.org/content/178/7/4517.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 30, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2007 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Evidence for Evolving Toll-IL-1 Receptor-Containing Adaptor Molecule Function in Vertebrates1 Con Sullivan,* John H. Postlethwait,† Christopher R. Lage,* Paul J. Millard,‡ and Carol H. Kim2* In mammals, Toll-IL-1R-containing adaptor molecule 1 (TICAM1)-dependent TLR pathways induce NF-B and IFN- re- sponses. -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Oas1b-Dependent Immune Transcriptional Profiles of West Nile
MULTIPARENTAL POPULATIONS Oas1b-dependent Immune Transcriptional Profiles of West Nile Virus Infection in the Collaborative Cross Richard Green,*,† Courtney Wilkins,*,† Sunil Thomas,*,† Aimee Sekine,*,† Duncan M. Hendrick,*,† Kathleen Voss,*,† Renee C. Ireton,*,† Michael Mooney,‡,§ Jennifer T. Go,*,† Gabrielle Choonoo,‡,§ Sophia Jeng,** Fernando Pardo-Manuel de Villena,††,‡‡ Martin T. Ferris,†† Shannon McWeeney,‡,§,** and Michael Gale Jr.*,†,1 *Department of Immunology and †Center for Innate Immunity and Immune Disease (CIIID), University of Washington, § Seattle, Washington 98109, ‡OHSU Knight Cancer Institute, Division of Bioinformatics and Computational Biology, Department of Medical Informatics and Clinical Epidemiology, and **Oregon Clinical and Translational Research Institute, Oregon Health & Science University, Portland, Oregon 97239, ††Department of Genetics and ‡‡Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, North Carolina 27514 ABSTRACT The oligoadenylate-synthetase (Oas) gene locus provides innate immune resistance to virus KEYWORDS infection. In mouse models, variation in the Oas1b gene influences host susceptibility to flavivirus infection. Oas However, the impact of Oas variation on overall innate immune programming and global gene expression flavivirus among tissues and in different genetic backgrounds has not been defined. We examined how Oas1b acts viral infection in spleen and brain tissue to limit West Nile virus (WNV) susceptibility and disease across a range of innate immunity genetic backgrounds. The laboratory founder strains of the mouse Collaborative Cross (CC) (A/J, C57BL/6J, multiparental 129S1/SvImJ, NOD/ShiLtJ, and NZO/HlLtJ) all encode a truncated, defective Oas1b, whereas the three populations wild-derived inbred founder strains (CAST/EiJ, PWK/PhJ, and WSB/EiJ) encode a full-length OAS1B pro- Multi-parent tein. -

Analysis of Chromatin Structure Indicates a Role for Fluoxetine in Altering Nucleosome Distribution Ely Gracia
Florida State University Libraries Honors Theses The Division of Undergraduate Studies 2012 Analysis of Chromatin Structure Indicates a Role for Fluoxetine in Altering Nucleosome Distribution Ely Gracia Follow this and additional works at the FSU Digital Library. For more information, please contact [email protected] THE FLORIDA STATE UNIVERSITY COLLEGE OF ARTS & SCIENCES ANALYSIS OF CHROMATIN STRUCTURE INDICATES A ROLE FOR FLUOXETINE IN ALTERING NUCLEOSOME DISTRIBUTION By: ELY GRACIA A Thesis submitted to the Department of Biological Sciences in partial fulfillment of the requirements for graduation with Honors in the Major Degree Awarded: Spring, 2012 1 The members of the Defense Committee approve the thesis of Ely Gracia defended on April 17, 2012. __________________________ Dr. Jonathan H. Dennis Thesis Director __________________________ Dr. Debra A. Fadool Committee Member __________________________ Dr. Joab Corey Outside Committee Member 2 Dedication: To my mother and father for all their love and support. To Nicole Cordero for always believing in me and pushing me to meet and exceed my potential. To Dr. Jonathan Dennis for allowing me the opportunity to challenge myself. 3 Abstract An interesting stimulus for chromatin structural changes is the generic and popular anti-depressant drug Fluoxetine, commonly known as Prozac. Generally accepted as a Selective Serotonin Reuptake Inhibitors (SSRI’s), recent work has emerged suggesting that this antidepressant also functions as a Histone Deaceylase Inhibitors (HDIs). Studies have also come out indicating that Fluoxetine acts as an immunosuppressant drug. Treatment with Fluoxetine is believed to reduce the over- activation of the immune system associated with depression. We have used an innovative microarray technology to measure changes in nucleosomal positioning that stem from Fluoxetine treatment. -

Microarray Analysis of Novel Genes Involved in HSV- 2 Infection
Microarray analysis of novel genes involved in HSV- 2 infection Hao Zhang Nanjing University of Chinese Medicine Tao Liu ( [email protected] ) Nanjing University of Chinese Medicine https://orcid.org/0000-0002-7654-2995 Research Article Keywords: HSV-2 infection,Microarray analysis,Histospecic gene expression Posted Date: May 12th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-517057/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/19 Abstract Background: Herpes simplex virus type 2 infects the body and becomes an incurable and recurring disease. The pathogenesis of HSV-2 infection is not completely clear. Methods: We analyze the GSE18527 dataset in the GEO database in this paper to obtain distinctively displayed genes(DDGs)in the total sequential RNA of the biopsies of normal and lesioned skin groups, healed skin and lesioned skin groups of genital herpes patients, respectively.The related data of 3 cases of normal skin group, 4 cases of lesioned group and 6 cases of healed group were analyzed.The histospecic gene analysis , functional enrichment and protein interaction network analysis of the differential genes were also performed, and the critical components were selected. Results: 40 up-regulated genes and 43 down-regulated genes were isolated by differential performance assay. Histospecic gene analysis of DDGs suggested that the most abundant system for gene expression was the skin, immune system and the nervous system.Through the construction of core gene combinations, protein interaction network analysis and selection of histospecic distribution genes, 17 associated genes were selected CXCL10,MX1,ISG15,IFIT1,IFIT3,IFIT2,OASL,ISG20,RSAD2,GBP1,IFI44L,DDX58,USP18,CXCL11,GBP5,GBP4 and CXCL9.The above genes are mainly located in the skin, immune system, nervous system and reproductive system. -

(12) United States Patent (10) Patent No.: US 7.873,482 B2 Stefanon Et Al
US007873482B2 (12) United States Patent (10) Patent No.: US 7.873,482 B2 Stefanon et al. (45) Date of Patent: Jan. 18, 2011 (54) DIAGNOSTIC SYSTEM FOR SELECTING 6,358,546 B1 3/2002 Bebiak et al. NUTRITION AND PHARMACOLOGICAL 6,493,641 B1 12/2002 Singh et al. PRODUCTS FOR ANIMALS 6,537,213 B2 3/2003 Dodds (76) Inventors: Bruno Stefanon, via Zilli, 51/A/3, Martignacco (IT) 33035: W. Jean Dodds, 938 Stanford St., Santa Monica, (Continued) CA (US) 90403 FOREIGN PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 WO WO99-67642 A2 12/1999 U.S.C. 154(b) by 158 days. (21)21) Appl. NoNo.: 12/316,8249 (Continued) (65) Prior Publication Data Swanson, et al., “Nutritional Genomics: Implication for Companion Animals'. The American Society for Nutritional Sciences, (2003).J. US 2010/O15301.6 A1 Jun. 17, 2010 Nutr. 133:3033-3040 (18 pages). (51) Int. Cl. (Continued) G06F 9/00 (2006.01) (52) U.S. Cl. ........................................................ 702/19 Primary Examiner—Edward Raymond (58) Field of Classification Search ................... 702/19 (74) Attorney, Agent, or Firm Greenberg Traurig, LLP 702/23, 182–185 See application file for complete search history. (57) ABSTRACT (56) References Cited An analysis of the profile of a non-human animal comprises: U.S. PATENT DOCUMENTS a) providing a genotypic database to the species of the non 3,995,019 A 1 1/1976 Jerome human animal Subject or a selected group of the species; b) 5,691,157 A 1 1/1997 Gong et al. -

Differentially Expressed Late Constituents of the Epidermal Cornified Envelope
Differentially expressed late constituents of the epidermal cornified envelope D. Marshall, M. J. Hardman, K. M. Nield, and C. Byrne* School of Biological Sciences, University of Manchester, 3.239 Stopford Building, Oxford Road, Manchester M13 9PT, United Kingdom Communicated by Elaine Fuchs, University of Chicago, Chicago, IL, September 17, 2001 (received for review June 23, 2001) Barrier activity of skin and internal barrier-forming epithelial There have been persistent reports of additional genes͞ linings are conferred by a lipid-corneocyte structure (stratum proteins with structures homologous to cornified envelope pro- corneum in skin).The integrity of the corneocytes depends on the teins. These include XP5, XP31, XP32 (12), the newly identified outer cornified envelope and is essential for maintenance of barrier component of the EDC (NICE-1; ref. 13), protein products of a function. During epidermal development and differentiation, pro- range of annotated expressed sequence tags (ESTs; ref. 14), and teins are sequentially incorporated into the envelope via action of Eig3 protein (15). We show that at least some of these proteins epidermal transglutaminases in a well documented process. How- are encoded by a previously undetected gene cluster in the ever, recent knockouts of major cornified envelope constituents human epidermal differentiation complex (EDC) at 1q21 (16), have failed to disrupt barrier function significantly, suggesting that with homologues detected in mouse (12, 14, 16). We show that additional unidentified components are involved. We report a new these genes encode proteins, which are new cornified envelope gene cluster in the epidermal differentiation complex at human constituents distinct from SPRRs. Like SPRRs, the genes are 1q21 encoding a family of 18 proteins that are substrates for differentially expressed in different types of barrier epithelia. -

Evaluation of a New Method for Large-Scale and Gene-Targeted Next Generation DNA Sequencing in Nonmodel Species
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 2013 Evaluation of a New Method for Large-Scale and Gene-targeted Next Generation DNA Sequencing in Nonmodel Species Ted Cosart The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Cosart, Ted, "Evaluation of a New Method for Large-Scale and Gene-targeted Next Generation DNA Sequencing in Nonmodel Species" (2013). Graduate Student Theses, Dissertations, & Professional Papers. 4133. https://scholarworks.umt.edu/etd/4133 This Dissertation is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. EVALUTATION OF A NEW METHOD FOR LARGE-SCALE AND GENE- TARGETED NEXT GENERATION DNA SEQUENCING IN NONMODEL SPECIES By Ted Cosart BA, University of Montana, Missoula, Montana, 1983 MS, University of Montana, Missoula, Montana, 2006 Dissertation presented in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Individualized, Interdisciplinary Graduate Program The University of Montana Missoula, Montana August, 2013 Approved by: Sandy Ross, Associate Dean of The Graduate School Graduate School Dr. Jesse Johnson, Co-Chair Computer Science Dr. Gordon Luikart, Co-Chair Flathead Biological Station Dr. Jeffrey Good Division of Biological Sciences Dr. William Holben Division of Biological Sciences Dr. Stephen Porcella Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases Dr. -

University of California, San Diego
UC San Diego UC San Diego Electronic Theses and Dissertations Title The post-terminal differentiation fate of RNAs revealed by next-generation sequencing Permalink https://escholarship.org/uc/item/7324r1rj Author Lefkowitz, Gloria Kuo Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs -

Evaluation on Reprogramed Biological Processes in Transgenic Maize
www.nature.com/scientificreports OPEN Evaluation on reprogramed biological processes in transgenic maize varieties using transcriptomics and metabolomics Wei Fu1, Pengyu Zhu1, Mingnan Qu2, Wang Zhi1, Yongjiang Zhang1, Feiwu Li3* & Shuifang Zhu1* Genetic engineering (GM) has great potential to improve maize productivity, but rises some concerns on unintended efects, and equivalent as their comparators. There are some limitations through targeted analysis to detect the UE in genetically modifed organisms in many previous studies. We here reported a case-study on the efects of introducing herbicides and insect resistance (HIR) gene cassette on molecular profling (transcripts and metabolites) in a popular maize variety Zhengdan958 (ZD958) in China. We found that introducing HIR gene cassette bring a limited numbers of diferential abundant genes (DAGs) or diferential abundant metabolites (DAMs) between transgenic events and non-transgenic control. In contrast, averaged 10 times more DAGs and DAMs were observed when performed comparison under diferent growing environments in three diferent ecological regions of China than the numbers induced by gene efects. Major biological pathways relating to stress response or signaling transduction could explain somehow the efects of growing environments. We further compared two transgenic events mediated ZD958 (GM-ZD958) with either transgenic parent GM-Z58, and other genetic background nonGM-Z58, nonGM-ZD958, and Chang7-2. We found that the numbers of DAGs and DAMs between GM-ZD958 and its one parent maize variety, Z58 or GM-Z58 is equivalent, but not Chang7-2. These fndings suggest that greater efects due to diferent genetic background on altered molecular profling than gene modifcation itself. This study provides a case evidence indicating marginal efects of gene pleiotropic efects, and environmental efects should be emphasized.