Syntheses and Evaluation of Putative Enzyme Inhibitor of Isoprenoid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

• Our Bodies Make All the Cholesterol We Need. • 85 % of Our Blood
• Our bodies make all the cholesterol we need. • 85 % of our blood cholesterol level is endogenous • 15 % = dietary from meat, poultry, fish, seafood and dairy products. • It's possible for some people to eat foods high in cholesterol and still have low blood cholesterol levels. • Likewise, it's possible to eat foods low in cholesterol and have a high blood cholesterol level SYNTHESIS OF CHOLESTEROL • LOCATION • All tissues • Liver • Cortex of adrenal gland • Gonads • Smooth endoplasmic reticulum Cholesterol biosynthesis and degradation • Diet: only found in animal fat • Biosynthesis: primarily synthesized in the liver from acetyl-coA; biosynthesis is inhibited by LDL uptake • Degradation: only occurs in the liver • Cholesterol is only synthesized by animals • Although de novo synthesis of cholesterol occurs in/ by almost all tissues in humans, the capacity is greatest in liver, intestine, adrenal cortex, and reproductive tissues, including ovaries, testes, and placenta. • Most de novo synthesis occurs in the liver, where cholesterol is synthesized from acetyl-CoA in the cytoplasm. • Biosynthesis in the liver accounts for approximately 10%, and in the intestines approximately 15%, of the amount produced each day. • Since cholesterol is not synthesized in plants; vegetables & fruits play a major role in low cholesterol diets. • As previously mentioned, cholesterol biosynthesis is necessary for membrane synthesis, and as a precursor for steroid synthesis including steroid hormone and vitamin D production, and bile acid synthesis, in the liver. • Slightly less than half of the cholesterol in the body derives from biosynthesis de novo. • Most cells derive their cholesterol from LDL or HDL, but some cholesterol may be synthesize: de novo. -

Rehmannia Glutinosa-Monocultured Rhizosphere Soil
Comparative Metaproteomic Analysis on Consecutively Rehmannia glutinosa-Monocultured Rhizosphere Soil Linkun Wu1,2, Haibin Wang1,2., Zhixing Zhang1,2., Rui Lin2,3, Zhongyi Zhang1,4, Wenxiong Lin1,2* 1 School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2 Agroecological Institute, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 3 College of Oceanography and Environmental Science, Xiamen University, Xiamen, Fujian, China, 4 Institute of Chinese Medicinal Materials, Henan Agriculture University, Zhengzhou, Henan, China Abstract Background: The consecutive monoculture for most of medicinal plants, such as Rehmannia glutinosa, results in a significant reduction in the yield and quality. There is an urgent need to study for the sustainable development of Chinese herbaceous medicine. Methodology/Principal Findings: Comparative metaproteomics of rhizosphere soil was developed and used to analyze the underlying mechanism of the consecutive monoculture problems of R. glutinosa. The 2D-gel patterns of protein spots for the soil samples showed a strong matrix dependency. Among the spots, 103 spots with high resolution and repeatability were randomly selected and successfully identified by MALDI TOF-TOF MS for a rhizosphere soil metaproteomic profile analysis. These proteins originating from plants and microorganisms play important roles in nutrient cycles and energy flow in rhizospheric soil ecosystem. They function in protein, nucleotide and secondary metabolisms, signal transduction and resistance. Comparative metaproteomics analysis revealed 33 differentially expressed protein spots in rhizosphere soil in response to increasing years of monoculture. Among them, plant proteins related to carbon and nitrogen metabolism and stress response, were mostly up-regulated except a down-regulated protein (glutathione S-transferase) involving detoxification. -
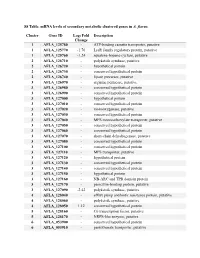
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

33 34 35 Lipid Synthesis Laptop
BI/CH 422/622 Liver cytosol ANABOLISM OUTLINE: Photosynthesis Carbohydrate Biosynthesis in Animals Biosynthesis of Fatty Acids and Lipids Fatty Acids Triacylglycerides contrasts Membrane lipids location & transport Glycerophospholipids Synthesis Sphingolipids acetyl-CoA carboxylase Isoprene lipids: fatty acid synthase Ketone Bodies ACP priming 4 steps Cholesterol Control of fatty acid metabolism isoprene synth. ACC Joining Reciprocal control of b-ox Cholesterol Synth. Diversification of fatty acids Fates Eicosanoids Cholesterol esters Bile acids Prostaglandins,Thromboxanes, Steroid Hormones and Leukotrienes Metabolism & transport Control ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Synthesis Degradation Ketone body Utilization Isoprene Biosynthesis 1 Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates are transferred stepwise from ATP to mevalonate. • A third phosphate from ATP is added at the hydroxyl, followed by decarboxylation and elimination catalyzed by pyrophospho- mevalonate decarboxylase creates a pyrophosphorylated 5-C product: D3-isopentyl pyrophosphate (IPP) (isoprene). • Isomerization to a second isoprene dimethylallylpyrophosphate (DMAPP) gives two activated isoprene IPP compounds that act as precursors for D3-isopentyl pyrophosphate Isopentyl-D-pyrophosphate all of the other lipids in this class isomerase DMAPP Cholesterol and Steroid Biosynthesis mevalonate kinase Mevalonate to Activated Isoprenes • Two phosphates -

Electric Supporting Information (ESI) Crystal Structure and Functional
Electronic Supplementary Material (ESI) for Chemical Science. This journal is © The Royal Society of Chemistry 2018 Electric Supporting Information (ESI) Crystal structure and functional analysis of large-terpene synthase belonging to a newly found subclass Masahiro Fujihashi,a* Tsutomu Sato,b* Yuma Tanaka,a Daisuke Yamamoto,a Tomoyuki Nishi,b Daijiro Ueda,b Mizuki Murakami,b Yoko Yasuno,c Ai Sekihara,c Kazuma Fuku,c Tetsuro Shinadac & Kunio Mikia* a. Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: [email protected], [email protected] b. Department of Applied Biological Chemistry, Faculty of Agriculture, and Graduate School of Science and Technology, Niigata University, 8050 Ikarashi-2, Niigata 950-2181, Japan. E-mail: [email protected] c. Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi, Osaka 558-8585, Japan. S1 Table of Contents Experimental Procedures S3 General procedure S3 Vector construction, expression and purification of BalTS S3 Crystallization and crystallographic analysis of balTS S3 Oligomeric state analysis S4 Analysis of the dimer geometry S4 Vector construction, expression and purification of BsuTS S4 Homology modeling of BsuTS S4 Enzymatic assays S5 Isolation and identification of β-springen S5 Movie Captions S6 Author Contributions S6 Figures Fig. S1 S7 Fig. S2 S8 Fig. S3 S9 Fig. S4 S10 Fig. S5 S17 Fig. S6 S18 Fig. S7, S8 S19 Fig. S9 S20 Fig. S10 S21 Fig. S11 S22 Tables Table S1 S23 Table S2 S24 References S27 S2 Experimental Procedures General procedure NMR spectra were recorded using a Bruker DPX 400 spectrometer at 400 MHz for proton (1H) and 100 MHz for carbon (13C). -

Steroid Interference with Antifungal Activity of Polyene Antibiotics
APPLIED MICROBIOLOGY, Nov., 1966 Vol. 14, No. 6 Copyright © 1966 American Society for Microbiology Printed in U.S.A. Steroid Interference with Antifungal Activity of Polyene Antibiotics WALTER A. ZYGMUNT AND PETER A. TAVORMINA Department of Microbiology and Natural Products Research, Mead Johnson & Company, Evansville, Indiana Received for publication 21 April 1966 ABSTRACT ZYGMUNT, WALTER A. (Mead Johnson & Co., Evansville, Ind.), AND PETER A. TAVORMINA. Steroid interference with antifungal activity of polyene antibiotics. Appl. Microbiol. 14:865-869. 1966.-Wide differences exist among the polyene antibiotics, nystatin, rimocidin, filipin, pimaricin, and amphotericin B, with ref- erence to steroid interference with their antifungal activities against Candida albicans. Of the numerous steroids tested, ergosterol was the only one which ef- fectively antagonized the antifungal activity of all five polyene antibiotics. The antifungal activities of nystatin and amphotericin B were the least subject to vitia- tion by the addition of steroids other than ergosterol, and those of filipin, rimo- cidin, and pimaricin were the most sensitive to interference. Attempts to delineate the structural requirements of steroids possessing polyene-neutralizing activity in growing cultures of C. albicans are discussed. The ultraviolet absorbance of certain antibiotic steroid combinations was also studied. It has been suggested (1, 9, 13) that the polyene While studying the effects of various steroids antibiotics become bound to the fungal cell mem- on the antimonilial activity of pimaricin, we brane and cause permeability changes with observed that ergostenol was almost as effective attendant depletion of essential cellular con- as the above A5-3/3-hydroxy steroids in antag- stituents. Loss of potassium and ammonium onizing pimaricin. -

BB 451/551 Lecture 35 Highlights
Kevin Ahern's Biochemistry (BB 451/551) at Oregon State University http://oregonstate.edu/instruct/bb451/summer13/lectures/highlightsglycer... Glycerolipid and Sphingolipid Metabolism 1. Phosphatidic acid is an immediate precursor of CDP-diacylglycerol, which is a precursor of the various glycerophospholipids . CTP combines with phosphatidic acid to yield a pyrophosphate and CDP-Diacylglycerol. Activation by CDP yields a high energy activated intermediate that can be readily converted to phosphatidyl glycerophospholipids. 2. From CDP-diacylglycerol, phosphatidyl serine can be made, as canphosphatidyl ethanolamine and phosphatidyl choline. Synthesis of phosphatidyl choline from phosphatidyl ethanolamine requires methyl groups donated by S-Adenoysyl-Methionine (SAM). Loss of the methyl groups by SAM yields S-Adenosyl-Homocysteine (I incorrectly said S-adenosyl-homoserine in the lecture). 3. Phosphatidyl ethanolamine (and phosphatidyl choline - derived from phosphatidyl ethanolamine) can both be made independently of phosphatidic acid biosynthesis. For this pathway, CDP-ethanolamine is the activated intermediate and the phosphoethanolamine of it is added to diacylglycerol to form phosphatidylethanolamine. Phosphatidyl choline can be made by the same methylation scheme in point 4. 4. Sphingolipids are synthesized beginning with palmitoyl-CoA and serine. Addition of a fatty acid to the amine group yields a ceramide. Addition of sugars to a ceramide yields either a cerebroside (single sugar added) or a ganglioside (complex sugar added). 5. Deficiencies in enzymes that degrade sphingolipids (particularly cerebrosides and gangliosides) are linked to neural disorders. One such disorder is Tay-Sachs disease. 6. Cholesterol is an important component of membranes, particularly in the brain. Cholesterol can be synthesized totally from acetyl-CoA. 7. Steroids include all compounds synthesized from cholesterol. -

Steroidal Triterpenes of Cholesterol Synthesis
Molecules 2013, 18, 4002-4017; doi:10.3390/molecules18044002 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Steroidal Triterpenes of Cholesterol Synthesis Jure Ačimovič and Damjana Rozman * Centre for Functional Genomics and Bio-Chips, Faculty of Medicine, Institute of Biochemistry, University of Ljubljana, Zaloška 4, Ljubljana SI-1000, Slovenia; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +386-1-543-7591; Fax: +386-1-543-7588. Received: 18 February 2013; in revised form: 19 March 2013 / Accepted: 27 March 2013 / Published: 4 April 2013 Abstract: Cholesterol synthesis is a ubiquitous and housekeeping metabolic pathway that leads to cholesterol, an essential structural component of mammalian cell membranes, required for proper membrane permeability and fluidity. The last part of the pathway involves steroidal triterpenes with cholestane ring structures. It starts by conversion of acyclic squalene into lanosterol, the first sterol intermediate of the pathway, followed by production of 20 structurally very similar steroidal triterpene molecules in over 11 complex enzyme reactions. Due to the structural similarities of sterol intermediates and the broad substrate specificity of the enzymes involved (especially sterol-Δ24-reductase; DHCR24) the exact sequence of the reactions between lanosterol and cholesterol remains undefined. This article reviews all hitherto known structures of post-squalene steroidal triterpenes of cholesterol synthesis, their biological roles and the enzymes responsible for their synthesis. Furthermore, it summarises kinetic parameters of enzymes (Vmax and Km) and sterol intermediate concentrations from various tissues. Due to the complexity of the post-squalene cholesterol synthesis pathway, future studies will require a comprehensive meta-analysis of the pathway to elucidate the exact reaction sequence in different tissues, physiological or disease conditions. -

Hydroxylation of 1-Deoxypentalenic Acid Mediated by CYP105D7 (SAV 7469) of Streptomyces Avermitilis
The Journal of Antibiotics (2011) 64, 65–71 & 2011 Japan Antibiotics Research Association All rights reserved 0021-8820/11 $32.00 www.nature.com/ja ORIGINAL ARTICLE Pentalenic acid is a shunt metabolite in the biosynthesis of the pentalenolactone family of metabolites: hydroxylation of 1-deoxypentalenic acid mediated by CYP105D7 (SAV_7469) of Streptomyces avermitilis Satoshi Takamatsu1,4, Lian-Hua Xu2,4, Shinya Fushinobu2, Hirofumi Shoun2, Mamoru Komatsu1, David E Cane3 and Haruo Ikeda1 Pentalenic acid (1) has been isolated from many Streptomyces sp. as a co-metabolite of the sesquiterpenoid antibiotic pentalenolactone and related natural products. We have previously reported the identification of a 13.4-kb gene cluster in the genome of Streptomyces avermitilis implicated in the biosynthesis of the pentalenolactone family of metabolites consisting of 13 open reading frames. Detailed molecular genetic and biochemical studies have revealed that at least seven genes are involved in the biosynthesis of the newly discovered metabolites, neopentalenoketolactone, but no gene specifically dedicated to the formation of pentalenic acid (1) was evident in the same gene cluster. The wild-type strain of S. avermitilis, as well as its derivatives, mainly produce pentalenic acid (1), together with neopentalenoketolactone (9). Disruption of the sav7469 gene encoding a cytochrome P450 (CYP105D7), members of which class are associated with the hydroxylation of many structurally different compounds, abolished the production of pentalenic acid (1). The sav7469-deletion mutant derived from SUKA11 carrying pKU462Hptl-clusterDptlH accumulated 1-deoxypentalenic acid (5), but not pentalenic acid (1). Reintroduction of an extra-copy of the sav7469 gene to SUKA11 Dsav7469 carrying pKU462Hptl-clusterDptlH restored the production of pentalenic acid (1). -

Antioxidant, Antimicrobial Effects and Phenolic Profile of Lycium Barbarum L
Molecules 2015, 20, 15060-15071; doi:10.3390/molecules200815060 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Article Antioxidant, Antimicrobial Effects and Phenolic Profile of Lycium barbarum L. Flowers Andrei Mocan 1, Laurian Vlase 2,*, Dan Cristian Vodnar 3, Ana-Maria Gheldiu 2, Radu Oprean 4 and Gianina Crișan 1 1 Department of Pharmaceutical Botany, Iuliu Hațieganu University of Medicine and Pharmacy, 23 Ghe. Marinescu Street, Cluj-Napoca 400010, Romania; E-Mails: [email protected] (A.M.); [email protected] (G.C.) 2 Department of Pharmaceutical Technology and Biopharmaceutics, Iuliu Hațieganu University of Medicine and Pharmacy, 12 I. Creangă Street, Cluj-Napoca 400010, Romania; E-Mail: [email protected] 3 Department of Food Science, University of Agricultural Sciences and Veterinary Medicine, 3-5 Manăştur Street, Cluj-Napoca 400372, Romania; E-Mail: [email protected] 4 Department of Analytical Chemistry and Instrumental Analysis, Iuliu Hațieganu University of Medicine and Pharmacy, 4 L. Pasteur Street, Cluj-Napoca 400010, Romania; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +40-264-595-770. Academic Editor: Milen I. Georgiev Received: 28 June 2015 / Accepted: 10 August 2015 / Published: 17 August 2015 Abstract: L. barbarum L. is a widely-accepted nutraceutical presenting highly advantageous nutritive and antioxidant properties. Its flowers have been previously described as a source of diosgenin, β-sitosterol and lanosterol that can be further pharmaceutically developed, but no other data regarding their composition is available. The purpose of this work was to investigate the chemical constituents, antioxidant and antimicrobial activities of L. -

Cholesterol, Phytosterols, Marine Sterols… 2
LIPIDS sterol lipids Marek Vecka CLASSIFICATION OF LIPIDS - molecular structure N of known Abbreviation Lipid class structures Fatty acyls FA 5869 Glycerolipids GL 7541 Glycerophospholipids GP 8002 Sphingolipids SP 4338 Sterol lipids ST 2715 Prenol lipids PL 1259 Other – saccharolipids, polyketides SL, PK 1293+6742 Fahy 2005, Fahy 2009 STEROL LIPIDS STEROL LIPIDS = lipid molecules with backbone derived from cyclopenta[a]phenanthrene (?) Division according to biochemical function 1. Sterols cholesterol, phytosterols, marine sterols… 2. Bile acids and derivatives C24, C26, C27, C28 bile acids, bile alcohols 3. Steroids C18 steroids, C19 steroids, C21 steroids 4.Secosteroids vitamins D Other groups conjugates, hopanoids, … STEROL LIPIDS Structures 1. Numbering system for C27 four-ring system first C´s on attached methyls side chain STEROL LIPIDS Structures 2. Stereochemistry Ring position b- a- substituent position Conventions: 1. Ring position: cis- (remaining 4th bonds of common C-C are cis-) (A-B cis-: bile acids) vs. trans- (remaining 4th bonds of common C-C are trans-) (all : cholesterol) 2. Substituents: a- (below cycle plane) vs. b- (above cycle plane) STEROL LIPIDS Structures 3. Important hydrocarbon structures C18 structures: estra- steroid hormones C19 structures: androsta- steroid hormones C21 structures: pregna- steroid hormones C24 structures: chola- bile acids/alcohols C27 structures: cholesta- cholesterol, oxysterols CLASSIFICATION OF LIPIDS - biosynthetic route Lipid class Biosynthetic route Fatty acyls condensation of thioesters Glycerolipids Glycerophospholipids Sphingolipids Sterol lipids condensation of activated isoprene units Prenol lipids Other – saccharolipids, polyketides other types STEROLS Biosynthesis of sterols (cholesterol) 1. Biosynthesis of isopentenyldiphosphate = activated isoprene unit 2. Condensation of isopentenyldiphosphate units 6 units are needed (C30) 3. -

(2020) Impact of PCSK9 Inhibition with Evolocumab on the Postprandial Responses of Triglyceride-Rich Lipoproteins in Type 2 Diabetic Subjects
Taskinen, M.-R. et al. (2020) Impact of PCSK9 inhibition with evolocumab on the postprandial responses of triglyceride-rich lipoproteins in type 2 diabetic subjects. Journal of Clinical Lipidology, 14(1), pp. 77-87. (doi: 10.1016/j.jacl.2019.12.003). This is the author’s final accepted version. There may be differences between this version and the published version. You are advised to consult the publisher’s version if you wish to cite from it. http://eprints.gla.ac.uk/207797/ Deposited on: 15 January 2020 Enlighten – Research publications by members of the University of Glasgow http://eprints.gla.ac.uk Impact of PCSK9 inhibition with evolocumab on the postprandial responses of triglyceride- rich lipoproteins in type 2 diabetic subjects Marja-Riitta Taskinen1, Elias Björnson2, Linda Andersson2, Juhani Kahri3, Kimmo Porthan1, Niina Matikainen1,4, Sanni Söderlund1,4, Kirsi Pietiläinen1,4, Antti Hakkarainen5, Nina Lundbom5, Ralf Nilsson6, Marcus Ståhlman2, Martin Adiels2, Paolo Parini7, Chris Packard8 and Jan Borén2,9 1Research Programs Unit, Clinical and Molecular Medicine, University of Helsinki, Helsinki, Finland; 2Department of Molecular and Clinical Medicine, Institute of Medicine, University of Gothenburg, Sweden; 3Department of Internal Medicine and Rehabilitation, Helsinki University Hospital, Helsinki, Finland; 4Endocrinology, Abdominal Center, Helsinki University Hospital, Helsinki, Finland; 5HUS Medical Imaging Center, Radiology, Helsinki University Hospital, University of Helsinki, Helsinki, Finland; 6Discovery Sciences, IMED