Evolutionary and Phylogenetic Aspects of the Chloroplast Genome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Botanical Gardens in France
France Total no. of Botanic Gardens recorded in France: 104, plus 10 in French Overseas Territories (French Guiana, Guadeloupe, Martinique and Réunion). Approx. no. of living plant accessions recorded in these botanic gardens: c.300,000 Approx. no. of taxa in these collections: 30,000 to 40,000 (20,000 to 25,000 spp.) Estimated % of pre-CBD collections: 80% to 90% Notes: In 1998 36 botanic gardens in France issued an Index Seminum. Most were sent internationally to between 200 and 1,000 other institutions. Location: ANDUZE Founded: 1850 Garden Name: La Bambouseraie (Maurice Negre Parc Exotique de Prafrance) Address: GENERARGUES, F-30140 ANDUZE Status: Private. Herbarium: Unknown. Ex situ Collections: World renowned collection of more than 100 species and varieties of bamboos grown in a 6 ha plot, including 59 spp.of Phyllostachys. Azaleas. No. of taxa: 260 taxa Rare & Endangered plants: bamboos. Special Conservation Collections: bamboos. Location: ANGERS Founded: 1895 Garden Name: Jardin Botanique de la Faculté de Pharmacie Address: Faculte Mixte de Medecine et Pharmacie, 16 Boulevard Daviers, F-49045 ANGERS. Status: Universiy Herbarium: No Ex situ Collections: Trees and shrubs (315 taxa), plants used for phytotherapy and other useful spp. (175 taxa), systematic plant collection (2,000 taxa), aromatic, perfume and spice plants (22 spp), greenhouse plants (250 spp.). No. of taxa: 2,700 Rare & Endangered plants: Unknown Location: ANGERS Founded: 1863 Garden Name: Arboretum Gaston Allard Address: Service des Espaces Verts de la Ville, Mairie d'Angers, BP 3527, 49035 ANGERS Cedex. Situated: 9, rue du Château d’Orgement 49000 ANGERS Status: Municipal Herbarium: Yes Approx. -

Garden Mastery Tips March 2006 from Clark County Master Gardeners
Garden Mastery Tips March 2006 from Clark County Master Gardeners Flowering Quince Flowering quince is a group of three hardy, deciduous shrubs: Chaenomeles cathayensis, Chaenomeles japonica, and Chaenomeles speciosa. Native to eastern Asia, flowering quince is related to the orchard quince (Cydonia oblonga), which is grown for its edible fruit, and the Chinese quince (Pseudocydonia sinensis). Flowering quince is often referred to as Japanese quince (this name correctly refers only to C. japonica). Japonica is often used regardless of species, and flowering quince is still called Japonica by gardeners all over the world. The most commonly cultivated are the hybrid C. superba and C. speciosa, not C. japonica. Popular cultivars include ‘Texas Scarlet,’ a 3-foot-tall plant with red blooms; ‘Cameo,’ a double, pinkish shrub to five feet tall; and ‘Jet Trail,’ a white shrub to 3 feet tall. Flowering quince is hardy to USDA Zone 4 and is a popular ornamental shrub in both Europe and North America. It is grown primarily for its bright flowers, which may be red, pink, orange, or white. The flowers are 1 to 2 inches in diameter, with five petals, and bloom in late winter or early spring. The glossy dark green leaves appear soon after flowering and turn yellow or red in autumn. The edible quince fruit is yellowish-green with reddish blush and speckled with small dots. The fruit is 2 to 4 inches in diameter, fragrant, and ripens in fall. The Good The beautiful blossoms of flowering quince Flowering quince is an easy-to-grow, drought-tolerant shrub that does well in shady spots as well as sun (although more sunlight will produce better flowers). -

Chaenomeles Speciosa) in the Naxi and Tibetan Highlands of NW Yunnan, China
Cultural and Ecosystem Services of Flowering Quince (Chaenomeles speciosa) in the Naxi and Tibetan Highlands of NW Yunnan, China. Authors: Lixin Yang, Selena Ahmed, John Richard Stepp, Yanqinag Zhao, Ma Jun Zeng, Shengji Pei, Dayuan Xue, and Gang Xu The final publication is available at Springer via https://dx.doi.org/10.1007/s12231-015-9318-7. Yang, Lixin, Selena Ahmed, John Richard Stepp, Yanqinag Zhao, Ma Jun Zeng, Shengji Pei, Dayuan Xue, and Gang Xu. “Cultural Uses, Ecosystem Services, and Nutrient Profile of Flowering Quince (Chaenomeles Speciosa) in the Highlands of Western Yunnan, China.” Economic Botany 69, no. 3 (September 2015): 273–283. doi:10.1007/s12231-015-9318-7. Made available through Montana State University’s ScholarWorks scholarworks.montana.edu Cultural Uses, Ecosystem Services, and Nutrient Profile Chaenomeles speciosa of Flowering Quince ( ) in the Highlands 1 of Western Yunnan, China 2,3 3,4 ,3,5 6 LIXIN YANG ,SELENA AHMED ,JOHN RICHARD STEPP* ,YANQINAG ZHAO , 7 2 ,3 2 MA JUN ZENG ,SHENGJI PEI ,DAYUAN XUE* , AND GANG XU 2State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institutes of Botany, Chinese Academy of Sciences, Kunming, China 3College of Life and Environmental Science, Minzu University of China, Beijing, China 4Department of Health and Human Development, Montana State University, Bozeman, MT, USA 5Department of Anthropology, University of Florida, Gainesville, FL, USA 6College of Forestry and Vocational Technology in Yunnan, Kunming, China 7Southwest Forestry University, Bailongshi, Kunming, China *Corresponding author; e-mail: [email protected]; [email protected] Introduction ample light but is tolerant of partial shade. -

List of the Import Prohibited Plants
List of the Import Prohibited Plants The Annexed Table 2 of the amended Enforcement Ordinance of the Plant Protection Law (Amended portions are under lined) Districts Prohibited Plants Quarantine Pests 1. Yemen, Israel, Saudi Arabia, Fresh fruits of akee, avocado, star berry, Mediterranean fruit fly Syria, Turkey, Jordan, Lebanon, allspice, olive, cashew nut, kiwi fruit, Thevetia (Ceratitis capitata) Albania, Italy, United Kingdom peruviana, carambola, pomegranate, jaboticaba, (Great Britain and Northern broad bean, alexandrian laurel, date palm, Ireland, hereinafter referred to as Muntingia calabura, feijoa, pawpaw, mammee "United Kingdom"), Austria, apple, longan, litchi, and plants of the genera Netherlands, Cyprus, Greece, Ficus, Phaseolus, Diospyros(excluding those Croatia, Kosovo, Switzerland, listed in appendix 41), Carissa, Juglans, Morus, Spain, Slovenia, Serbia, Germany, Coccoloba, Coffea, Ribes, Vaccinium, Hungary, France, Belgium, Passiflora, Dovyalis, Ziziphus, Spondias, Musa Bosnia and Herzegovina, (excluding immature banana), Carica (excluding Portugal, Former Yugoslav those listed in appendix 1), Psidium, Artocarpus, Republic of Macedonia, Malta, , Annona, Malpighia, Santalum, Garcinia, Vitis Montenegro, Africa, Bermuda, (excluding those listed in appendices 3 and 54), Argentina, Uruguay, Ecuador, El Eugenia, Mangifera (excluding those listed in Salvador, Guatemala, Costa Rica, appendices 2 ,36 ,43 ,51 and 53), Ilex, Colombia, Nicaragua, West Indies Terminalia and Gossypium, and Plants of the (excluding Cuba, Dominican family Sapotaceae, Cucurbitaceae (excluding Republic,Puerto Rico), Panama, those listed in appendices 3 and 42), Cactaceae Paraguay, Brazil, Venezuela, (excluding those listed in appendix 35), Peru, Bolivia, Honduras, Australia Solanaceae (excluding those listed in (excluding Tasmania), Hawaiian appendices 3 and 42), Rosaceae (excluding Islands those listed in appendices 3 and 31) and Rutaceae (excluding those listed in appendices 4 to 8 ,39 ,45 and 56). -

Evolution of Angiosperm Pollen. 7. Nitrogen-Fixing Clade1
Evolution of Angiosperm Pollen. 7. Nitrogen-Fixing Clade1 Authors: Jiang, Wei, He, Hua-Jie, Lu, Lu, Burgess, Kevin S., Wang, Hong, et. al. Source: Annals of the Missouri Botanical Garden, 104(2) : 171-229 Published By: Missouri Botanical Garden Press URL: https://doi.org/10.3417/2019337 BioOne Complete (complete.BioOne.org) is a full-text database of 200 subscribed and open-access titles in the biological, ecological, and environmental sciences published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Complete website, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/terms-of-use. Usage of BioOne Complete content is strictly limited to personal, educational, and non - commercial use. Commercial inquiries or rights and permissions requests should be directed to the individual publisher as copyright holder. BioOne sees sustainable scholarly publishing as an inherently collaborative enterprise connecting authors, nonprofit publishers, academic institutions, research libraries, and research funders in the common goal of maximizing access to critical research. Downloaded From: https://bioone.org/journals/Annals-of-the-Missouri-Botanical-Garden on 01 Apr 2020 Terms of Use: https://bioone.org/terms-of-use Access provided by Kunming Institute of Botany, CAS Volume 104 Annals Number 2 of the R 2019 Missouri Botanical Garden EVOLUTION OF ANGIOSPERM Wei Jiang,2,3,7 Hua-Jie He,4,7 Lu Lu,2,5 POLLEN. 7. NITROGEN-FIXING Kevin S. Burgess,6 Hong Wang,2* and 2,4 CLADE1 De-Zhu Li * ABSTRACT Nitrogen-fixing symbiosis in root nodules is known in only 10 families, which are distributed among a clade of four orders and delimited as the nitrogen-fixing clade. -

Phylogeny of Maleae (Rosaceae) Based on Multiple Chloroplast Regions: Implications to Genera Circumscription
Hindawi BioMed Research International Volume 2018, Article ID 7627191, 10 pages https://doi.org/10.1155/2018/7627191 Research Article Phylogeny of Maleae (Rosaceae) Based on Multiple Chloroplast Regions: Implications to Genera Circumscription Jiahui Sun ,1,2 Shuo Shi ,1,2,3 Jinlu Li,1,4 Jing Yu,1 Ling Wang,4 Xueying Yang,5 Ling Guo ,6 and Shiliang Zhou 1,2 1 State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing 100093, China 2University of the Chinese Academy of Sciences, Beijing 100043, China 3College of Life Science, Hebei Normal University, Shijiazhuang 050024, China 4Te Department of Landscape Architecture, Northeast Forestry University, Harbin 150040, China 5Key Laboratory of Forensic Genetics, Institute of Forensic Science, Ministry of Public Security, Beijing 100038, China 6Beijing Botanical Garden, Beijing 100093, China Correspondence should be addressed to Ling Guo; [email protected] and Shiliang Zhou; [email protected] Received 21 September 2017; Revised 11 December 2017; Accepted 2 January 2018; Published 19 March 2018 Academic Editor: Fengjie Sun Copyright © 2018 Jiahui Sun et al. Tis is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Maleae consists of economically and ecologically important plants. However, there are considerable disputes on generic circumscription due to the lack of a reliable phylogeny at generic level. In this study, molecular phylogeny of 35 generally accepted genera in Maleae is established using 15 chloroplast regions. Gillenia isthemostbasalcladeofMaleae,followedbyKageneckia + Lindleya, Vauquelinia, and a typical radiation clade, the core Maleae, suggesting that the proposal of four subtribes is reasonable. -
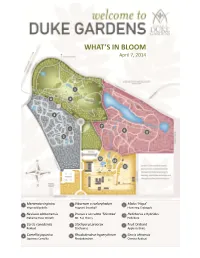
What's in Bloom
WHAT’S IN BLOOM April 7, 2014 5 4 6 2 7 1 9 8 3 12 10 11 1 Mertensia virginica 5 Viburnum x carlcephalum 9 Malus ‘Hopa’ Virginia Bluebells Fragrant Snowball Flowering Crabapple 2 Neviusia alabamensis 6 Prunus x serrulata ‘Shirotae’ 10 Helleborus x hybridus Alabama Snow Wreath Mt. Fuji Cherry Hellebore 3 Cercis canadensis 7 Stachyurus praecox 11 Fruit Orchard Redbud Stachyurus Apple cultivars 4 Camellia japonica 8 Rhododendron hyperythrum 12 Cercis chinensis Japanese Camellia Rhododendron Chinese Redbud WHAT’S IN BLOOM April 7, 2014 BLOMQUIST GARDEN OF NATIVE PLANTS Amelanchier arborea Common Serviceberry Sanguinaria canadensis Bloodroot Cornus florida Flowering Dogwood Stylophorum diphyllum Celandine Poppy Thalictrum thalictroides Rue Anemone Fothergilla major Fothergilla Trillium decipiens Chattahoochee River Trillium Hepatica nobilis Hepatica Trillium grandiflorum White Trillium Hexastylis virginica Wild Ginger Hexastylis minor Wild Ginger Trillium pusillum Dwarf Wakerobin Illicium floridanum Florida Anise Tree Trillium stamineum Blue Ridge Wakerobin Malus coronaria Sweet Crabapple Uvularia sessilifolia Sessileleaf Bellwort Mertensia virginica Virginia Bluebells Pachysandra procumbens Allegheny spurge Prunus americana American Plum DORIS DUKE CENTER GARDENS Camellia japonica Japanese Camellia Pulmonaria ‘Diana Clare’ Lungwort Cercis canadensis Redbud Prunus persica Flowering Peach Puschkinia scilloides Striped Squill Cercis chinensis Redbud Sanguinaria canadensis Bloodroot Clematis armandii Evergreen Clematis Spiraea prunifolia Bridalwreath -

The Origin of Alternation of Generations in Land Plants
Theoriginof alternation of generations inlandplants: afocuson matrotrophy andhexose transport Linda K.E.Graham and LeeW .Wilcox Department of Botany,University of Wisconsin, 430Lincoln Drive, Madison,WI 53706, USA (lkgraham@facsta¡.wisc .edu ) Alifehistory involving alternation of two developmentally associated, multicellular generations (sporophyteand gametophyte) is anautapomorphy of embryophytes (bryophytes + vascularplants) . Microfossil dataindicate that Mid ^Late Ordovicianland plants possessed such alifecycle, and that the originof alternationof generationspreceded this date.Molecular phylogenetic data unambiguously relate charophyceangreen algae to the ancestryof monophyletic embryophytes, and identify bryophytes as early-divergentland plants. Comparison of reproduction in charophyceans and bryophytes suggests that the followingstages occurredduring evolutionary origin of embryophytic alternation of generations: (i) originof oogamy;(ii) retention ofeggsand zygotes on the parentalthallus; (iii) originof matrotrophy (regulatedtransfer ofnutritional and morphogenetic solutes fromparental cells tothe nextgeneration); (iv)origin of a multicellularsporophyte generation ;and(v) origin of non-£ agellate, walled spores. Oogamy,egg/zygoteretention andmatrotrophy characterize at least some moderncharophyceans, and arepostulated to represent pre-adaptativefeatures inherited byembryophytes from ancestral charophyceans.Matrotrophy is hypothesizedto have preceded originof the multicellularsporophytes of plants,and to represent acritical innovation.Molecular -

Download Download
Natural Barrens and Post Oak Flatwoods in Posey and Spencer Counties, Indiana James R. Aldrich and Michael A. Homoya Indiana Natural Heritage Program, Indiana Department of Natural Resources Indianapolis, Indiana 46204 Introduction Post oak flatwoods are xeric forested communities that are dominated by Quercus stellata with a relatively open canopy that allows a great deal of dispersed light to reach the forest floor. This forested community lacks the typical shrub-layer found in more mesic forested communities and appears "savanna-like". Characteristically, post oak flatwoods in Indiana are found on poorly drained nearly level soils of alluvial lacustrine terraces of the Ohio River and other major streams in the unglaciated region of the Wabash Lowland Physiographic Province (12). The understory is dominated by sedges and the content of organic matter in the soil appears to be very low. The barrens described herein are relatively small natural openings surrounded by post oak flatwoods where a fragipan is at or very near the surface. The vegetation in these bar- rens is not dominated by prairie grasses and forbs, but is closely related to the vegeta- tion of sandstone glades described for Illinois (21) and Missouri (18). Contemporary southern "flatwoods" of the Illinois Tillplain dominated by sweetgum {Liquidambar styraciflua), beech (Fagus grandifolia) and red maple (Acer rubrum) in southwestern Ohio (4) and southeastern Indiana have been described in detail (9,13,15,17). Indiana "barrens" dominated by prairie forbs and grasses (7) have been discovered recently and discussed (3,10). Flatwoods dominated by post oak have received limited attention in Illinois (16) where they have been referred to as "southern flatwoods" (21) and very little has been written about the Indiana post oak flatwoods. -

Cell Wall Ribosomes Nucleus Chloroplast Cytoplasm
Cell Wall Ribosomes Nucleus Nickname: Protector Nickname: Protein Maker Nickname: Brain The cell wall is the outer covering of a Plant cell. It is Ribosomes read the recipe from the The nucleus is the largest organelle in a cell. The a strong and stiff and made of DNA and use this recipe to make nucleus directs all activity in the cell. It also controls cellulose. It supports and protects the plant cell by proteins. The nucleus tells the the growth and reproduction of the cell. holding it upright. It ribosomes which proteins to make. In humans, the nucleus contains 46 chromosomes allows water, oxygen and carbon dioxide to pass in out They are found in both plant and which are the instructions for all the activities in your of plant cell. animal cells. In a cell they can be found cell and body. floating around in the cytoplasm or attached to the endoplasmic reticulum. Chloroplast Cytoplasm Endoplasmic Reticulum Nickname: Oven Nickname: Gel Nickname: Highway Chloroplasts are oval structures that that contain a green Cytoplasm is the gel like fluid inside a The endoplasmic reticulum (ER) is the transportation pigment called chlorophyll. This allows plants to make cell. The organelles are floating around in center for the cell. The ER is like the conveyor belt, you their own food through the process of photosynthesis. this fluid. would see at a supermarket, except instead of moving your groceries it moves proteins from one part of the cell Chloroplasts are necessary for photosynthesis, the food to another. The Endoplasmic Reticulum looks like a making process, to occur. -

Chaenomeles Spp. - Flowering Quince (Rosaceae) ------Chaenomeles Is a Functional Flowering Hedge, Border, Twigs Or Specimen Shrub That Can Be Used Near Buildings
Chaenomeles spp. - Flowering Quince (Rosaceae) ----------------------------------------------------------------------------------- Chaenomeles is a functional flowering hedge, border, Twigs or specimen shrub that can be used near buildings. -buds small and reddish in color The major appeal of Flowering Quince is its showy -lightly armed (terminal and axillary spines) but brief flowering period. The rest of the year it’s a -young bark is reddish and cherry-like utilitarian thorny shrub with limited aesthetic Trunk attributes. -gray brown -many small diameter stems closely crowded, arising FEATURES from the ground Form -large shrubs USAGE 2-6' tall Function -vased shaped -sun tolerant, long-lived shrub habit with -useful as a hedge or barrier many small Texture diameter -medium in foliage and when bare stems Assets -1:1 height to -urban tolerant width ratio -withstand severe pruning -rapid growth -drought tolerant Culture -early spring flowers -full sun -dense growth and long-lived -adaptable to a wide range of soil conditions -lightly armed for effective "crowd control" -thrives under stressful conditions Liabilities -moderate availability -poor autumn color Foliage -trash can accumulate among its many small diameter -alternate, lanceolate stems (maintenance headache) -serrate margins -prone to cosmetic damage by insects -somewhat leathery -sheds foliage in summer in response to drought or -to 4" long disease pressure -leafy, kidney-shaped stipules (an ID feature) Habitat -summer color is dense medium green and attractive, -Zones 4 to 8, depending on species new growth often bronze -Native to the Orient (China, Japan) -autumn color yellowish green SELECTIONS Alternates -urban tolerant shrub with vase-shaped winter form (e.g. Berberis thunbergii, Berberis x mentorensis, Spiraea nipponica 'Snowmound' etc.) -early spring flowering shrubs (e.g. -

Brown Algae and 4) the Oomycetes (Water Molds)
Protista Classification Excavata The kingdom Protista (in the five kingdom system) contains mostly unicellular eukaryotes. This taxonomic grouping is polyphyletic and based only Alveolates on cellular structure and life styles not on any molecular evidence. Using molecular biology and detailed comparison of cell structure, scientists are now beginning to see evolutionary SAR Stramenopila history in the protists. The ongoing changes in the protest phylogeny are rapidly changing with each new piece of evidence. The following classification suggests 4 “supergroups” within the Rhizaria original Protista kingdom and the taxonomy is still being worked out. This lab is looking at one current hypothesis shown on the right. Some of the organisms are grouped together because Archaeplastida of very strong support and others are controversial. It is important to focus on the characteristics of each clade which explains why they are grouped together. This lab will only look at the groups that Amoebozoans were once included in the Protista kingdom and the other groups (higher plants, fungi, and animals) will be Unikonta examined in future labs. Opisthokonts Protista Classification Excavata Starting with the four “Supergroups”, we will divide the rest into different levels called clades. A Clade is defined as a group of Alveolates biological taxa (as species) that includes all descendants of one common ancestor. Too simplify this process, we have included a cladogram we will be using throughout the SAR Stramenopila course. We will divide or expand parts of the cladogram to emphasize evolutionary relationships. For the protists, we will divide Rhizaria the supergroups into smaller clades assigning them artificial numbers (clade1, clade2, clade3) to establish a grouping at a specific level.