10 Things Packaging Engineers Should Know
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cardboard and Brown Paper Bags Office Paper, Newspaper, Junk Mail, Magazines, and Catalogs
Recycling Center 801 Diamond Valley Drive Open: Daily to the public during daylight hours This guide will help you properly prepare your recyclable materials for drop-off at the Town of Windsor Recycle Center. This is a drop-off facility. It does not have a buy-back option and is for use by residents and small businesses. Following this information will help maintain the facility and the recycling program for the benefit of the community. IMPORTANT… • Do not leave your recyclables in plastic bags. Plastic bags are NOT recyclable! • The plastic item must be a BOTTLE or JAR. with a #1 or #2 on the bottom. • 99 percent of these will have a screw-on plastic lid (which isn’t recyclable). • Plastic containers with a #3 - #7 on the bottom are NOT acceptable. • Tubs, buckets, deli plates, microwave/fast food trays, wrappers, Styrofoam, toys, patio furniture, etc. are NOT acceptable. • Plastic bottles larger than 2.5 gallons are NOT acceptable. • Syringes and other medical supplies are NOT acceptable. Cardboard and Brown Paper Bags Corrugated cardboard is easy to recognize. It is made of paper and has an arched layer called “fluting” between smooth sheets called “liners”. The drop-off site has two 40-yard hydraulic compactor units for collecting corrugated cardboard and brown paper bags. The compaction system is self-activated by depositing the prepared materials into a six-inch tall slot. Flatten boxes. Cut or tear large boxes into sections no larger than 4 feet by 4 feet to prevent jamming the machine. No wet, waxed-coated or food-contaminated boxes. -
Tyvek Graphics Technical Data Sheet
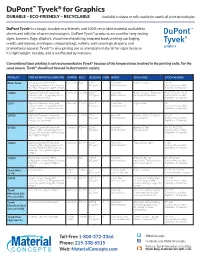
DuPont ™ Tyvek ® for Graphics DURABLE – ECO-FRIENDLY – RECYCLABLE Available in sheets or rolls; usable for nearly all print technolo gies DuPont Tyvek a is a tough, durable, eco-friendly and 100% recyclable material available in sheets and rolls for all print technologies. DuPont Tyvek ® products are used for long-lasting signs, banners, flags, displays, visual merchandising, map and book printing, packaging, credit card sleeves, envelopes, shopping bags, wallets, wall coverings, drapery, and promotional apparel. Tyvek ® is also gaining use as a template material for signs because it is lightweight, durable, and is unaffected by moisture. Conventional laser printing is not recommended on Tyvek ® because of the temperatures involved in the printing units. For the same reason, Tyvek ® should not be used in electrostatic copiers. PRODUCT TYPE OF PRINTING USED FOR COATED MILS OZ. [GSM] CORE NOTES IDEAL USES STOCK WIDTHS Black Tyvek Flexography, Gravure, Offset Uncoated 5 mil 1.25 oz 2” Paper-like. Banners & Signs 36", 45" Lithography, Screen Process, UV-cure [42 gsm] Hard Structure Custom sizes available Inkjet (w/ testing due to lighter weight) Available in 10-yard rolls 1085D Digital on Demand, Flexography, Uncoated 10.3 mil 3.2 oz 3” Paper-like. Banners & Signs. Extra body 48.25", 57.125", 114.25" Gravure, Offset Lithography, Screen [109 gsm] Hard Structure for shape development. Custom sizes available Process, UV-cure Inkjet Available in 10-yard rolls 1079 Digital on Demand, Flexography, Uncoated 7.9 mil 2.85 oz 3” Paper-like. Tags & Labels 48" Gravure, Offset Lithography, Screen [97 gsm] Hard Structure Custom sizes available Process, Thermal Transfer, UV-cure Available in 10-yard rolls Inkjet 1073D Digital on Demand, Flexography, Uncoated 7.5 mil 2.2 oz 3” Paper-like. -

Getinge Pack Tyvek Sterilization Reels and Pouches
SAFETY DATA SHEET Getinge Pack Tyvek Sterilization Reels and Pouches 1.0 Product Product Name Getinge Pack Tyvek Sterilization Reels and Pouches and Company Product Code Tyvek Reel (TY)- Tyvek Pouch (TP) Identification Intended Use Tyvek Sterilization Reels and Pouches are intended to be used to enclose another medical devices that are to be sterilized by health care provider via related sterilization methods. Supplier PMS Tıbbi Cihazlar Teknolojisi Sanayi ve Ticaret A.Ş Identification Address Karaduvar Mah. Serbest Bölge 11. Cadde No: 46,Akdeniz, 33020 Mersin/TURKEY Phone +90 324 238 70 42 Fax +90 324 238 65 49 2.0 Hazards Tyvek material and PET/PE laminated film are not classified as dangerous according to EC directives and other international safety guidelines. Identification 3.0 Composition Information on Ingredients • Tyvek Material (1059B, 64.4 g/m² ) / Information on • Polyethylene Terephthalate / Polyethylene (PET/PE) Laminated Film Ingredients 12/50 microns • Printing inks, Indicator inks 4.0 First Aid Eye contact If molten material or dust contacts the eye, immediatly flush Measures with plenty of water for at least 15 minutes. If easy to do , remove contact lenses. Get medical attention immediately. Skin contact If contact with molten product occurs, treat as for thermal burn. Cool melted product on skin with plenty of water. Do not remove solidified product. If dust contacts the skin, immediately flush with plenty of water. Inhalation Remove patient to fresh air. Seek medical attention if necessary. Ingestion Material is not expected to be absorbed from the gastrointestinal tract. However, the printing chemicals such as printing inks and chemical indicator inks could be harmful. -

1610 8 Shashoua Icomcc 2017
ICOM-CC 18th Triennial Conference Sustainable future alternatives 2017 Copenhagen to petroleum-based polymeric SCIENTIFIC RESEARCH conservation materials YVONNE SHASHOUA* INTRODUCTION National Museum of Denmark Kongens Lyngby, Denmark [email protected] Conservation treatments often employ petroleum-based plastic materials KATJA JANKOVA ATANASOVA as packaging, adhesives and coatings that are synthesised from non- Technical University of Denmark Kongens Lyngby, Denmark renewable crude oil, a resource at risk of exhaustion within the next [email protected] 100 years. The Going Green conference held at the British Museum in CLAIRE CURRAN ICA Art Conservation April 2009 concluded that conservators are under increasing pressure to Cleveland OH, USA [email protected] review their practices in light of international environmental targets and *Author for correspondence the rising costs of fossil fuels. Biopolymers are considered sustainable either because they are synthesised from renewable sources or because they biodegrade to CO2 and H2O in soil and water after use. The range and KEYWORDS: sustainable, biopolymer, bioplastic quality of bioplastics have increased dramatically since 2006 and, today, polyethylene, polyester, soya, humic acid polyethylenes, polyesters, polyurethanes and polyvinyl alcohols can be fully synthesised from biomass, although their commercial availability ABSTRACT is more limited in Europe than in the USA and South America. While The research described here is the first study extensive research has been conducted into the rates and mechanism of on the use of sustainable, plant-based biopoly- degradation of bioplastics on disposal (Rani et al. 2012), few projects have mers in conservation practice. Two applications of biopolymers to conservation were investi- focused on their chemical and physical properties during use and none gated – in commercial bioplastics as substi- have addressed the application of bioplastics to conservation practice. -

Coa Ng Conver Ng Distribu
s! t Last Tha ond A B Self-Adhesive Tapes Electrical Insula� on Tapes Industrial Tapes Packaging Tapes Double Sided Tapes Die-Cut Solu� ons Specialty Tapes Dispensers & Processing Devices Coa� ng Conver� ng Distribu� on Volz Selbstklebetechnik GmbH YOUR EXPERT IN ADHESIVE TAPE SOLUTIONS VOLZ® TAPES is your competent partner for all self-adhesive tape requirements. Our extensive product por� olio offers a variety of standard and specialty tape products, suppor� ng advanced industry applica� ons, as well as standard and complex packaging require- ments. We are the industry expert in double-sided tapes, electrical and industrial tapes, packaging tapes, self-adhesive die-cut solu� ons, as well as tape dispensers and processing devices. VOLZ® TAPES has spent the last several decades developing its core competencies in the areas of Coa� ng, Conver� ng and Distribu� on. We produce turn-key custom tape solu� ons to meet our customers’ most complex industry challenges. Our advanced produc� on facility is capable of cu� ng any tape width or producing die-cut solu� ons to your exact specifica� ons. It is our goal to develop high-end, cost effec� ve tape solu� ons that meet your opera� onal demands. VOLZ® TAPES is an ac� ve and trusted member of AFERA, the European Adhesive Tape Associa� on. AFERA membership provides a front row seat to market developments and valuable exposure to other key adhesive tape associa� ons such as PSTC (USA), CATIA (China), TAAT (Taiwan) and JATMA (Japan). • Coa� ng of diverse carrier materials such as Fabric, PP, PE, PET, PVC, Fleece and COATING: CUSTOMIZED ADHESIVE SOLUTIONS Foam VOLZ® TAPES develops and manufactures high-end electrical and • Available Adhesives Include: double-sided adhesive tapes. -

Tyvek ® Printing Guide
, China 兽桃 Mask Bag, designed by Shou Tao Tao Mask Bag, designed by Shou DuPont™ Tyvek ® Graphics EMEA Printability Guide Water Resistant Paper-like Light Tear Resistant Recyclable Printable DuPont™ Tyvek ® Graphics EMEA Printability Guide DuPont™ Tyvek® is a popular printing substrate due to its light weight, smooth surface, high dimensional stability, opacity, toughness and durability. Uncoated Tyvek® can be printed using most digital and commercial printing processes. Some digital presses and some aqueous ink jet printers require a special coating. Tyvek® can be printed either sheet or web-fed. Tyvek® can be printed the same way as paper, although some of its physical properties do require special attention. To achieve excellent print quality, both the designer and printer must understand the unique properties and characteristics of Tyvek®. Tyvek® is made of continuous high-density polyethylene filaments. By using heat and pressure, these filaments are bonded into a base material for printing which turns out to be neither paper, cloth nor plastic film, but it integrates the advantages of those three materials. Tyvek® material has a melting point of 135°C and is a water-resistant and non-absorbent material with superior dimensional stability, high strength, and a smooth matt surface. Most traditional printing technologies can be used for Tyvek® printing, as well as some digital printing. The following Tyvek® printing quick reference guidelines have been summarized based on our current knowledge and the relevant contents will be updated -

Dupont™ Tyvek® Water-Resistive Barriers Installation Guidelines
DuPont™ Tyvek® Water-Resistive Barriers Installation Guidelines HELPING YOU GET THE JOB DONE RIGHT VERSION 2 Table of Contents Applicable Products ..................................................................................................................................................................2 Recommended Materials .........................................................................................................................................................2 Code Requirements ..................................................................................................................................................................3 General Instructions .................................................................................................................................................................3 Special Considerations .............................................................................................................................................................3 Installation Instructions .............................................................................................................................................................4 Continuity Terminations ........................................................................................................................................................................6 Gable Ends ...........................................................................................................................................................................6 -

Supply Chain Packaging Guide
Secondary Packaging Supply Chain Standards July 7, 2021 Business Confidential | ©2021 Walmart Stores, Inc. 177 // 338 Secondary Packaging Supply Chain Standards - Update Summary These standards have included multiple clarifications of what is required and what is NOT ALLOWED. These changes have been updated throughout the published standards to provide clarity to suppliers. The pages have been reorganized to provide a better flow. PAGE 2021 UPDATES Changes to Supply Chain Standards 185 SQEP Phase 2 and Phase 3 Defect Description/Definitions Added 202 General Case Markings Updated for Dates, Unprocessed Meats, and Cylindrical Items 210-213 Updated Pallet Standards 218 Update "Palletized Shipments" to "Unitized Shipments" 227 Add Inbound Appointment Scheduling Standard 228 Update TV Test Standards 235-237 Add Direct Store Delivery (DSD) aka Direct To Store (DTS) Standards 239 Update SIOC Standards 240 Add eCommerce Product Specific Requirement Standards 241-244 Add Drop Ship Vendor (DSV) Standards 268 Add Jewelry Distribution Center Standards 269-271 Add Optical Distribution Center Standards 275 Add Goods Not For Resale (GNFR) Standards 277-278 Update Meat/Poultry/Seafood Case and Pallet Label Standards 284 Add HACCP Pallet Placard for GCC Shipments 311-312 Add Frozen Seafood Carton Marking Requirements Appendix D Update Receiving Pulp Temperature Range Business Confidential | © 2021 Walmart Stores, Inc. The examples shown are for reference only. Supply Chain Standards 178 // 338 Table of Contents Supply Chain Stretch Wrap . 219 Produce Shipments . 280 Contact Information . 179 Trailer Loading . 220 Automated Grocery Handling . 281 Walmart Retail Link Resources . 180 Trailer Measurements. 221 Grocery Import Distribution Center (GIDC) . 282 Walmart Distribution Center Overview . -

(Cgmp) for Drugs
Questions and Answers on Current Good Manufacturing Practices, Goo... http://www.fda.gov/cder/guidance/cGMPs/packaging.htm FDA Home Page | CDER Home Page | CDER Site Info | Contact CDER | What's New @ CDER Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance Packaging and Labeling Control 1. Do CGMPs require that forced degradation studies always be conducted of the drug product when determining if a drug product stability test method is stability- indicating? 2. Can containers, closures, and packaging materials be sampled for receipt examination in the warehouse? 1. Do CGMPs require that forced degradation studies always be conducted of the drug product when determining if a drug product stability test method is stability- indicating? No. Drug product stress testing (forced degradation) may not be necessary when the routes of degradation and the suitability of the analytical procedures can be determined through use of the following: data from stress testing of drug substance reference materials for process impurities and degradants data from accelerated and long-term studies on drug substance data from accelerated and long-term studies on drug product Additional supportive information on the specificity of the analytical methods and on degradation pathways of the drug substance may be available from literature sources. Section 211.165(e) of the CGMP regulations states that the accuracy, sensitivity, specificity, and reproducibility of test methods shall be established and documented. Further, section 211.166(a)(3) requires that stability test methods be reliable, meaningful, and specific, which means that the content of active ingredient, degradation products, and other components of interest in a drug product can be accurately measured without interference, often called "stability-indicating." The CGMP regulations do not specify what techniques or tests are to be used to ensure that one’s test methods are stability-indicating. -

Dupont Tyvek Brochure
™ Optimize the Building Envelope DuPont DuPont™ ® DuPont Building Envelope systems help make buildings more DuPont has been the industry leader since it invented the Tyvek durable, comfortable, and energy-efficient. building wrap category more than 30 years ago with the RoofLiner HomeWrap® introduction of DuPont™ Tyvek® HomeWrap®. Today, we’re Whether it’s a skyscraper or a single-family home, the building working with architects, builders and installers on innovative envelope is an essential line of defense against air, water and solutions to protect the next generation of new construction wasted energy. and renovation of existing buildings. DuPont Building Envelope systems offer solutions that meet or DuPont™ exceed codes, help extend building life, and help reduce fossil fuel consumption. They resist moisture and air, but are highly StraightFlash™ VF permeable, to reduce the risk of condensation damage, wood rot or mold growth. DuPont™ Tyvek® StuccoWrap® DuPont™ StraightFlash™ DuPont™ Flashing Tape DuPont™ Tyvek® DrainWrap™ DuPont™ Tyvek® Tape DuPont™ DuPont™ FlexWrap™ NF Tyvek® ThermaWrap™ R5.0 DuPont™ Residential Additional Products Available: Sealant DuPont™ DuPont™ ™ ™ Adhesive/Primer RainVent Batten DuPont DuPont™ ® Tyvek DuPont™ DuPont™ Tyvek® Wrap Caps ThermaWrap™ LE Window and Door Foam Tyvek Fluid Applied System DuPont™ DuPont™ Tyvek® HomeWrap® SUPPORT WHEN AND RoofLiner e world’s leading house wrap has the optimum combination of A lightweight, easy-to-use roong underlayment that oers properties to deliver the best balance of weather protection, WHERE YOU NEED IT superior tear and UV resistance with improved water hold-out moisture management and durability behind residential facades. DuPont Building Knowledge Center™ ALL THE PRODUCTS and better mold resistance than traditional roong felt. -

Dupont Tyvek Homewrap Physical Properties Data Sheet
Product Information Sheet DuPont™ Tyvek® HomeWrap™ Sturdy House Wrap Engineered for Energy Efficiency FEATURES/BENEFITS Description Air and Water Barrier Performance DuPont™ Tyvek® HomeWrap™ is the original house wrap, The unique nonwoven structure of Tyvek® HomeWrap™ makes it incorporating unique material science that helps keep air and breathable, allowing moisture vapor to pass through. This helps water out, while letting water vapor escape. As a result, it can promote drying in wall systems, to help prevent mold and water contribute to improved building durability by helping to damage. In addition, Tyvek® HomeWrap™ stops air movement protect homes against damaging wind and rain that can through the walls, helping insulation perform closer to its full penetrate the exterior cladding. R-value, to provide a more energy-efficient home. Tyvek® HomeWrap™ can also reduce home energy bills by Ease of Installation controlling air flow and water intrusion, which helps insulation Tyvek® HomeWrap™ is easy to install. It is pliable, so it wraps work better, allowing the HVAC system to work more around corners with ease. It is also light weight, easier to handle, efficiently. It’s a house wrap engineered to keep homes cool in and faster to install than the average house wrap. In addition, the summer, warm in the winter, and dry all year round. because it’s flexible, Tyvek® HomeWrap™ easily interfaces at joints, and over architectural elements. Available Sizes Tyvek® HomeWrap™ is available in 9- and 10-foot width rolls for use behind a variety of claddings. This width minimizes seams and offers the potential for reduction in labor costs, compared to narrower rolls. -

The Office of Cannabis Regulation Packaging and Labeling Guide for Medical Marijuana Version 1 March 2020
State of Rhode Island and Providence Plantations DEPARTMENT OF BUSINESS REGULATION Office of Cannabis Regulation 1511 Pontiac Avenue, Bldg. 68-1 Cranston, Rhode Island 02920 The Office of Cannabis Regulation Packaging and Labeling Guide for Medical Marijuana Version 1 March 2020 This document is meant to help explain the packaging and labeling rules effective on March 25, 2020. However, this guide should not replace a thorough reading of the rules found here: https://rules.sos.ri.gov/regulations/part/230-80-05-1. 1 Table of Contents Product Type Page Number Flower Packaging 3 Labeling 4 Concentrate Packaging and Device 6 Labeling 7 Edible Single Serving- Solid 9 Multiple Single Serving- Solid 10 Single Serving- Liquid 11 Multiple Single Serving- Liquid 12 Bundled 13 Labeling 14 Ingestible Single Serving- Solid 16 Multiple Single Serving- Solid 17 Single Serving- Liquid 18 Multiple Single Serving- Liquid 19 Bundled 20 Labeling 21 Child-Resistant 23 Exit Packaging 24 Universal Symbol Stamping 25 2 Flower Packaging Requirements Packaging must be: • Opaque. • Light-Resistant. • Tamper- Evident. • Neutral in Color. • Certified as Child-Resistant (see page 24 for additional requirements). • Resealable. Confirm the Packaging: • Protects the product from contamination. • Does not impart any toxic or deleterious substance to the product. • Fully encloses the product. • Does not contain any design, image, label or coloring that was not approved or required by the OCR. 3 Flower Labeling Requirements Font Requirements for required information: • No smaller than size 6 font. • Permitted Font Types: Times New Roman, Calibri, Arial, Helvetica. • Must be in Black or White. • Clearly printed in the English language.