Structural Effects on Enzymatic Activity of Bacteriophage T4 Lysozyme Upon Adsorption to Colloidal Silica
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

I I I I Executive Summary
I I I CONTAMINANTS IN FISH AND SEDIMENTS OF THE GREAT SWAMP NATIONAL WILDLIFE REFUGE, MORRIS COUNTY, NEW JERSEY: I A 10-YEAR FOLLOW-UP INVESTIGATION DEC ID# 9950003.1 I ECDM Catalog # 5050048 I I I Prepared for: U.S. Fish & Wildlife Service Northeast Region Division of Refuges and Wildlife I Hadley, Massachusetts I Prepared by: I U.S. Fish & Wildlife Service New Jersey Field Office I Plem,antville, New Jersey I I August 2005 I Project Biologist: Clay M. Stem Assistant Supervisor: Timothy J. Kubiak I Supen,isor: Clifford G. Day I I 2 I I I I EXECUTIVE SUMMARY Located in Morris County, New Jersey about 25 miles west of New York City's Time Square, the I U.S. Fish & Wildlife Service's (Service) Great Swamp National Wildlife Refuge (GSNWR) encompasses approximately 7,500 acres ::,fwhich 3,660 acres are designated and managed as a National Wilderness Area. The GSNWR's wetlands provide important ecological functions, I including floodwater attenuation, groundwater recharge, pollution abatement, wildlife habitat, as well as recreational benefits for the public. In many portions of the Great Swamp watershed, significant areas of native soils have been disturbed by development to the extent that the I original soil profiles no longer exist. Development-related activities such as grading, infilling, and compaction have adversely altered the native soil's infiltration capacity and runoff potential I and thereby have increased storm-water 11ediment loading into many of the watershed's streams. The purpose of this investigation is to conduct a 10-year follow-up to a 1988 investigation (USFWS 1991) characterizing ambient concentrations of metals, organochlorines, and polycyclic I aromatic hydrocarbons (P AHs) in GSNWR sediments, and metals and organochlorine in fish inhabiting the GSNWR. -
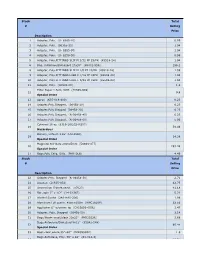
Stock Total # Selling Price 1 Adapter, Poly. (U- 6365-10)
Stock Total # Selling Price Description 1 Adapter, Poly. (U- 6365-10) 0.99 2 Adapter, Poly. (06365-22) 1.04 3 Adapter, Poly. (U- 6365-30) 1.04 4 Adapter, Poly. (U- 6359-50) 0.99 5 Adapter, Poly,FITTINGS SLIP M 3/32 PP 25/PK (45518-24) 1.04 6 Bag, Autoclave/Biohazard 24x30'' (95042-556) 156.1 8 Adapter, Poly.FITTINGS SLIP M 1/8 PP 25/PK (45518-26) 1.59 9 Adapter, Poly.FITTINGS LUER F 1/16 PP 25PK (45508-00) 1.04 10 Adapter, Poly.FITTINGS LUER F 3/32 PP 25PK (45508-02) 1.04 11 Adapter, Poly. (30800-08) 1.3 Filter Paper 1.5cm, VWR (28309-989) 12 9.9 Special Order 13 Apron (635-018-400) 0.23 14 Adapter,Poly.Stepped, (06458-10) 6.23 15 Adapter,Poly.Stepped (06458-20) 6.23 16 Adapter,Poly.Stepped, (N-06458-40) 6.23 17 Adapter,Poly.Stepped, (N-06458-60) 5.06 Cytoseal 16 oz (8310-16/23244257) 18 58.49 Hazardous Battery, Lithium 3.6V (LS14500) 19 14.38 Special Order Magnetic Stir Bars 2mmx5mm (58948-377) 20 293.48 Special Order 21 Bags,Poly Cello,10 lb. (PKR10Lb) 4.49 Stock Total # Selling Price Description 22 Adapter,Poly. Stepped (N-06458-30) 2.71 24 Alconox (21835-032) 42.75 25 Ammonium Sulfate,certif. (A7023) 41.18 26 Bar, spin 2'' x 1/2'' (14-51367) 5.22 27 Alcohol Swabs (248-HAS-200) 1.96 28 Aluminum Foil comm. 45cmx100m (WPC1810P) 32.56 29 Applicator 6'' w/cotton tip (CA10806-000L) 1.47 30 Adapter, Poly. -

Digestion of Algin by Pseudomonas Maltophilia and Pseudomonas Putida V
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Jan. 1980, p. 92-96 Vol. 39, No. 1 0099-2240/80/01-0092/05$02.00/0 Digestion of Algin by Pseudomonas maltophilia and Pseudomonas putida V. LYLE VON RIESEN Department ofMedical Microbiology, College ofMedicine, University of Nebraska Medical Center, Omaha, Nebraska 68105 Pseudomonas maltophilia and Pseudomonas putida were identified as al- ginolytic species. Two media used for demonstrating alginolytic activity are described. The applied aspects of the ability of these two species to digest algin are discussed. In a search for undescribed biochemical activ- Both media contained yeast extract (0.5%), algin (al- ities of nonfastidious, nonfermentative gram- ginic acid, sodium salt, no. A-7128; Sigma Chemical negative bacilli (NFB) which might be used to Co., St. Louis, Mo.) (1%), and bromothymol blue of these taxonomically (0.002%). Agar (0.5%) was used to form an algin-agar aid in the identification medium and animal charcoal (0.1%) to form an algin- troublesome organisms, a survey of a variety of charcoal medium. The media were prepared in 100- to complex polysaccharides as substrates was made 500-ml amounts in the following manner. Calculated by using a few strains of some well-recognized amounts of yeast extract and bromothymol blue (from species of NFB. When it was discovered that a 1% aqueous solution) were added to an appropriate some of the strains ofPseudomonas maltophilia volume of distilled water. This was then warmed on a showed evidence of an ability to hydrolyze algin magnetic stirrer-heater. When warm (40 to 60°C), (sodium alginate), an available collection of ap- algin and charcoal (algin-charcoal medium) or algin proximately 390 strains of NFB was examined and agar (algin-agar medium) were sprinkled into the of individual strains to digest vigorously stirred solution. -

Colonie\Project Plans\VP Soils Work Plan\VP-WP-Final.Wpd
VP Soils Work Plan for the Colonie FUSRAP Site Vicinity Properties By: U. S. Army Corps of Engineers New York District 26 Federal Plaza, Room 1811 New York, New York 10278 (917) 790-8230 Report No. 2008012/G-202374 August 2, 2011 VP Soils Work Plan for the Colonie FUSRAP Site Vicinity Properties By: U. S. Army Corps of Engineers New York District 26 Federal Plaza, Room 1811 New York, New York 10278 (917) 790-8230 Contractor: Integrated Environmental Management, Inc. 975 Russell Avenue, Suite A Gaithersburg, Maryland 20879 (240) 631-8990 Report No. 2008012/G-202374 August 2, 2011 U. S. ARMY CORPS OF ENGINEERS - NEW YORK DISTRICT “VP Soils Work Plan for the Colonie FUSRAP Site Vicinity Properties" Contract No. W912DR-08-D-0022, Delivery Order 002 August 2, 2011 FINAL Page i of 49 TABLE OF CONTENTS ACRONYMS AND ABBREVIATIONS ..........................................1 1 PROJECT DESCRIPTION ..................................................2 1.1 Introduction .........................................................2 1.2 Facility Description ...................................................2 1.3 Facility History ......................................................2 1.4 Contaminant Identification .............................................2 1.5 VP Background ......................................................3 1.5.1 Property at 50 Yardboro Avenue .................................3 1.5.2 Property at 1118 Central Avenue .................................3 1.6 Project Objectives ....................................................3 2 PROJECT -

Bio 215 Course Title: General Biochemistry Laboratory I
NATIONAL OPEN UNIVERSITY OF NIGERIA SCHOOL OF SCIENCE AND TECHNOLOGY COURSE CODE: BIO 215 COURSE TITLE: GENERAL BIOCHEMISTRY LABORATORY I 1 BIO 215: GENERAL BIOCHEMISTRY LABORATORY I Course Developer: DR (MRS.) AKIN-OSANAIYE BUKOLA CATHERINE UNIVERISTY OF ABUJA Course Editor: Dr Ahmadu Anthony Programme Leader: Professor A. Adebanjo Course Coordinator: Adams Abiodun E. 2 UNIT 1 INTRODUCTION TO LABORATORY AND LABORATORY EQUIPMENT CONTENTS 1.0 Introduction 2.0 Objectives 3.0 Main Body 3.1 What is a Laboratory? 3.2 Laboratory Equipment 4.0 Conclusion 5.0 Summary 6.0 Tutor-marked Assignments 7.0 References/Further Readings 1.0 INTRODUCTION Scientific research and investigations will be of little value without good field and laboratory work. These investigations are normally carried out through the active use of processes which involves laboratory or other hands-on activities. Many devices and products used in everyday life resulted from laboratory works. They include car engines, plastics, radios, televisions, synthetic fabrics, etc. 3 2.0 OBJECTIVES Upon completion of studying this unit, you should be able to: 1. Define the meaning of laboratory 2. List different types of laboratories 3. Identify different types of laboratory equipment 3.0 MAIN BODY 3.1 What is a Laboratory? A laboratory informally, lab is a facility that provides controlled conditions in which scientific research, experiments, and measurement may be performed. It is a place equipped for investigative procedures and for the preparation of reagents, therapeutic chemical materials, and so on. The title of laboratory is also used for certain other facilities where the processes or equipment used are similar to those in scientific laboratories. -

Pharmaceutical Arena
Journal of Automatic Chemistry, Vol. 14, No. 2 (March-April 1992), pp. 37-41 Managing robotics in the generic pharmaceutical arena Marianne Scheffler Danbury Pharmacal, Inc., 12 Stoneleigh Avenue, Carmel, New York 10512, USA Robotics was introduced by Danbury Pharmacal, Inc. in 1987 in order to improve laboratory throughput for several new products. The author uses Danbury Pharmacal's experience to give an overview ofvarious issues, such as acceptance by senior management and chemists, political confrontation, validation and product throughput. Introduction The pharmaceutical industry, both generic (multi- source), as well as PMA ('Brand') firms have a primary obligation to provide to the user (patient) finished dosage forms which meet all mandated standards for identity, purity, strength, and quality. A key difference between a generic pharmaceutical or multisource company and a major PMA firm are the larger number and greater variety of products manufactured by the generic firm. The pharmaceutical industry continues to be challenged with stricter government regulations, which has placed A) Hand G H) Flack #3 Test Tube Rack on B) Liquld/Uquid Station I) Hand O increasing demands firms for better and faster C) Rack #1. Fleaker Rack J) Ra #4 Test Tube Rack analytical testing capabilities. Pressure to meet produc- D) Ooital Shaker K) D & D E) Hind C L) Spe SIP tion schedules without compromising product quality F) Rack #2 Fleaker Rack M) Balance often becomes the driving force to improve laboratory G) Fleakar Capng Station Off The Pie 1. Diode An'ay Speclropholomelar productivity. Improving laboratory productivity is com- 2. MLS Slallon shorter with the 3. -

Soil Survey Laboratory Methods Manual Is to Document Methodology and to Serve As a Reference for the Laboratory Analyst
United States Department of Agriculture Natural Resources Conservation Service Soil Survey Laboratory Methods Manual Soil Survey Investigations Report No. 42 Version 4.0 November 2004 SOIL SURVEY LABORATORY METHODS MANUAL Rebecca Burt, Editor Soil Survey Investigations Report No. 42 Version 4.0 November 2004 Cover Photo:The cover illustrates the continuum of data collected or produced for the soil survey of a geographic area. The soil survey map, landscape, and pedon are from northern Iredell County, North Carolina and are representative of the Banister pedon. This series is classified as a fine, mixed, active, mesic Aquic Hapludalf. This soil is a very deep, moderately to somewhat poorly drained soil on stream terrace positions in the Piedmont physiographic province of Virginia and North Carolina. Laboratory data sheet illustrates particle-size data produced by the Soil Survey Laboratory. The photomicrograph of soil fabric features a slightly altered, exfoliated biotite grain under cross-polarized light. Bar scale in photo is 0.25 mm. (Photo credit of Banister landscape and pedon: John Kelly and Michael A. Wilson, NRCS, Raleigh, NC and NRCS, Lincoln, NE.) FOREWORD Laboratory data are critical to the understanding of the properties and genesis of a single pedon, as well as to the understanding of fundamental soil relationships based on many observations of a large number of soils. Development of both an analytical database and the soil relationships based on those data are the cumulative effort of generations of soil scientists at the Soil Survey Laboratory (SSL). The purpose of the Soil Survey Investigations Report (SSIR) No., Soil Survey Laboratory Methods Manual is to document methodology and to serve as a reference for the laboratory analyst. -

Trade Catalogs Trade Catalogs
Trade Catalogs Trade Catalogs Trade Literature and Advertisements Collection by Manufacturer To request copies of trade literature and advertisements, please email the Curator at [email protected]. We also have a manuals collection. NUMERIC | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z NUMERIC 10X Genomics 2015 Revolutionizing Gene Expression with Single-Cell RNAseq 3M, St. Paul, MN: 1966 Scotch-Grip Industrial Adhesives Product Line 1966 Industrial Adhesive no. 847 1966 Industrial Adhesive no. 226 1979 3M Display Film 1982 Information: Fluorel Fluoroelastomers 1983 Fluorel Fluoroelastomers: New Freedom for the Design Engineer 1986 Guide to Evaluation: Petrifilm Plates 1980s Nextel 312 Ceramic Fiber from 3M 1980s Ad: The energy-saving insulation of the future is available in the shape you need today 1980s Ad: When Dr. Hal Sowman told us Nextel 312 ceramic fiber could handle 2600F, we thought he was out to lunch. 1980s Nextel 312: How Nextel 312 Ceramic Fiber compares with the next best high temperature fiber 1980s Nextel 312: Product Bulletin: Braided Sleeving, Hose Coverings & Tapes 1980s Nextel 312: Product Bulletin: Furnace Curtains 1980s Nextel 312: Product Bulletin: Insulation for heating elements for preheating and stress relief applications 1980s Nextel 312: Product Bulletin: Vacuum Furnace Linings 1983 Nextel Ceramic Fiber for High Temperature Applications Where Dependability Counts 1983 Nextel Ceramic Fiber Products: For High Temperature Applications 1983 Nextel 312 Woven Tape Product Bulletin 1983 Nextel 312 Woven Fabrics 1983 Nextel Sewing Thread 1983 Nextel Health Safety Bulletin 1983 Nextel 312 Braided Sleeving top of page A A-1 Plating, Baltimore, MD: 1986, Military Plating Specifications ABSciex, Framingham, MA: 2010 MPX-2 System 2011 A New Dimension in Selectivity 2012 The Accurate Mass Workhorse Ace Glass Inc.: 1982 Ace Bulletin Vol. -

Review of Particle Size Distribution Analysis Methods
REVIEW OF PARTICLE SIZE DISTRIBUTION ANALYSIS METHODS JORGE GUEVARA ADVISOR: L.R. ELLIS University of Florida Gainesville, FL INTRODUCTION Particle size distribution analysis (PSDA) is a measurement of the size distribution of individual soil/sediment particles, sand, silt and clay, which can be used to understand soil genesis, to classify soil or to define texture (Soil Survey Staff, 1993). The USDA classification of soil texture is based on the proportion of sand 2.0-0.05 mm, silt 0.05-0.002 mm and clay < 0.002 mm particles (Table 1). Geologists and sedimentologists have been on the forefront of analyzing marine and submerged sediments. Demas was one of the first to take a soil science approach to study the sediments of shallow coastal systems (Demas et al, 1996). A soil science approach to sediments would necessitate the classification of these subaqueous soils into a unified taxonomic system with the Natural Resource Conservation Service’s (NRCS) terrestrial soil taxonomy (Soil Survey Staff, 1999). Thomas Reinsch of the NRCS’ National Soil Survey Center explained that PSDA data will be collected for subaqueous soils in regards to existing policies and methods with the terrestrial soils (personal communication with Dr. Thomas Reinsch). PSDA is a major criterion used to describe these soils and their characteristics. PSDA of subaqueous soils will be consistent with the terrestrial standards of the established NRCS methods. The current procedure accepted by the NRCS for PSDA is the pipette method (Gee and Bauder, 1986). The USDA uses this method because it is reproducible on many different types of soils (Soil Survey Staff, 1999). -

Laboratory Labware Organizers Drawer/Ganizers® and Refrig/Arrangers®
K03E Laboratory Labware Organizers Drawer/Ganizers® and Refrig/Arrangers® Drawer/Ganizers® Spacers are designed to maximize laboratory working space while increasing safety. The durable, light-weight polystyrene Spacers fit in standard size lab drawers with separate compartment designations that organize and protect supplies commonly found in the lab, as well as reduce breaks and spills that pose potential hazards in the laboratory setting. Protect supplies Conserve space Increase handling efficiency Transport Promote safety Go to Drawer/Ganizer® Organizer Trays Refrig/Arranger® Storage Trays Watch a video Drawer/Ganizer® Portable Organizer Trays Spectrum introduces new Drawer/Ganizer Portable Trays molded from 1/8 inch thick polystyrene plastic with easy gripping folded edges for transporting assorted lab items and mobilizing experimental activities. The deeper compartments are better suited than most other lab trays for holding larger quantities as well as serving as secondary containment for spills. Like the original Drawer/Ganizer Drawer Spacers, these Portable Organizer Trays are designed to fit standard lab drawers, refrigerators and bench tops to organize and protect supplies, conserve valuable space and promote lab safety. Spectrum offers 8 functional configurations (1 tray/pkg). Organize & Protect Lab Supplies Designed for Portability & Storage Stronger & More Durable Promotes Lab Safety Provides Secondary Containment 9 Portable Organizer Tray Configurations: Pipette / Column Tray Stopper / Fitting Tray Parts / Chrom Tray Two sizes for sorting 4 or 6 types of Great for separating large quantities of Various size compartments for easy to pipettes, chromatography columns or long different types of small items. 2 sizes find storage of lab & chromatography instruments in a protected parallel available with 12 or 16 equal supplies like utensils, valves, connectors, position. -

1455189355674.Pdf
THE STORYTeller’S THESAURUS FANTASY, HISTORY, AND HORROR JAMES M. WARD AND ANNE K. BROWN Cover by: Peter Bradley LEGAL PAGE: Every effort has been made not to make use of proprietary or copyrighted materi- al. Any mention of actual commercial products in this book does not constitute an endorsement. www.trolllord.com www.chenaultandgraypublishing.com Email:[email protected] Printed in U.S.A © 2013 Chenault & Gray Publishing, LLC. All Rights Reserved. Storyteller’s Thesaurus Trademark of Cheanult & Gray Publishing. All Rights Reserved. Chenault & Gray Publishing, Troll Lord Games logos are Trademark of Chenault & Gray Publishing. All Rights Reserved. TABLE OF CONTENTS THE STORYTeller’S THESAURUS 1 FANTASY, HISTORY, AND HORROR 1 JAMES M. WARD AND ANNE K. BROWN 1 INTRODUCTION 8 WHAT MAKES THIS BOOK DIFFERENT 8 THE STORYTeller’s RESPONSIBILITY: RESEARCH 9 WHAT THIS BOOK DOES NOT CONTAIN 9 A WHISPER OF ENCOURAGEMENT 10 CHAPTER 1: CHARACTER BUILDING 11 GENDER 11 AGE 11 PHYSICAL AttRIBUTES 11 SIZE AND BODY TYPE 11 FACIAL FEATURES 12 HAIR 13 SPECIES 13 PERSONALITY 14 PHOBIAS 15 OCCUPATIONS 17 ADVENTURERS 17 CIVILIANS 18 ORGANIZATIONS 21 CHAPTER 2: CLOTHING 22 STYLES OF DRESS 22 CLOTHING PIECES 22 CLOTHING CONSTRUCTION 24 CHAPTER 3: ARCHITECTURE AND PROPERTY 25 ARCHITECTURAL STYLES AND ELEMENTS 25 BUILDING MATERIALS 26 PROPERTY TYPES 26 SPECIALTY ANATOMY 29 CHAPTER 4: FURNISHINGS 30 CHAPTER 5: EQUIPMENT AND TOOLS 31 ADVENTurer’S GEAR 31 GENERAL EQUIPMENT AND TOOLS 31 2 THE STORYTeller’s Thesaurus KITCHEN EQUIPMENT 35 LINENS 36 MUSICAL INSTRUMENTS -

Determination of Ideal Broth Formulations Needed to Prepare Hydrous Hafnium Oxide Microspheres Via the Internal Gelation Process
ORNL/TM-2006/123 Determination of Ideal Broth Formulations Needed to Prepare Hydrous Hafnium Oxide Microspheres via the Internal Gelation Process December 2008 J. L. Collins R. D. Hunt S. G. Simmerman DOCUMENT AVAILABILITY Reports produced after January 1, 1996, are generally available free via the U.S. Department of Energy (DOE) Information Bridge: Web site: http://www.osti.gov/bridge Reports produced before January 1, 1996, may be purchased by members of the public from the following source: National Technical Information Service 5285 Port Royal Road Springfield, VA 22161 Telephone: 703-605-6000 (1-800-553-6847) TDD: 703-487-4639 Fax: 703-605-6900 E-mail: [email protected] Web site: http://www.ntis.gov/support/ordernowabout.htm Reports are available to DOE employees, DOE contractors, Energy Technology Data Exchange (ETDE) representatives, and International Nuclear Information System (INIS) representatives from the following source: Office of Scientific and Technical Information P.O. Box 62 Oak Ridge, TN 37831 Telephone: 865-576-8401 Fax: 865-576-5728 E-mail: [email protected] Web site: http://www.osti.gov/contact.html This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof.