GFP Plasmid) 35X
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cloning of Gene Coding Glyceraldehyde-3-Phosphate Dehydrogenase Using Puc18 Vector
Available online a t www.pelagiaresearchlibrary.com Pelagia Research Library European Journal of Experimental Biology, 2015, 5(3):52-57 ISSN: 2248 –9215 CODEN (USA): EJEBAU Cloning of gene coding glyceraldehyde-3-phosphate dehydrogenase using puc18 vector Manoj Kumar Dooda, Akhilesh Kushwaha *, Aquib Hasan and Manish Kushwaha Institute of Transgene Life Sciences, Lucknow (U.P), India _____________________________________________________________________________________________ ABSTRACT The term recombinant DNA technology, DNA cloning, molecular cloning, or gene cloning all refers to the same process. Gene cloning is a set of experimental methods in molecular biology and useful in many areas of research and for biomedical applications. It is the production of exact copies (clones) of a particular gene or DNA sequence using genetic engineering techniques. cDNA is synthesized by using template RNA isolated from blood sample (human). GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) is one of the most commonly used housekeeping genes used in comparisons of gene expression data. Amplify the gene (GAPDH) using primer forward and reverse with the sequence of 5’-TGATGACATCAAGAAGGTGGTGAA-3’ and 5’-TCCTTGGAGGCCATGTGGGCCAT- 3’.pUC18 high copy cloning vector for replication in E. coli, suitable for “blue-white screening” technique and cleaved with the help of SmaI restriction enzyme. Modern cloning vectors include selectable markers (most frequently antibiotic resistant marker) that allow only cells in which the vector but necessarily the insert has been transfected to grow. Additionally the cloning vectors may contain color selection markers which provide blue/white screening (i.e. alpha complementation) on X- Gal and IPTG containing medium. Keywords: RNA isolation; TRIzol method; Gene cloning; Blue/white screening; Agarose gel electrophoresis. -

Molecular Biology and Applied Genetics
MOLECULAR BIOLOGY AND APPLIED GENETICS FOR Medical Laboratory Technology Students Upgraded Lecture Note Series Mohammed Awole Adem Jimma University MOLECULAR BIOLOGY AND APPLIED GENETICS For Medical Laboratory Technician Students Lecture Note Series Mohammed Awole Adem Upgraded - 2006 In collaboration with The Carter Center (EPHTI) and The Federal Democratic Republic of Ethiopia Ministry of Education and Ministry of Health Jimma University PREFACE The problem faced today in the learning and teaching of Applied Genetics and Molecular Biology for laboratory technologists in universities, colleges andhealth institutions primarily from the unavailability of textbooks that focus on the needs of Ethiopian students. This lecture note has been prepared with the primary aim of alleviating the problems encountered in the teaching of Medical Applied Genetics and Molecular Biology course and in minimizing discrepancies prevailing among the different teaching and training health institutions. It can also be used in teaching any introductory course on medical Applied Genetics and Molecular Biology and as a reference material. This lecture note is specifically designed for medical laboratory technologists, and includes only those areas of molecular cell biology and Applied Genetics relevant to degree-level understanding of modern laboratory technology. Since genetics is prerequisite course to molecular biology, the lecture note starts with Genetics i followed by Molecular Biology. It provides students with molecular background to enable them to understand and critically analyze recent advances in laboratory sciences. Finally, it contains a glossary, which summarizes important terminologies used in the text. Each chapter begins by specific learning objectives and at the end of each chapter review questions are also included. -

Pspark® DNA Cloning System Manual for Cat
pSpark® DNA cloning system Manual for cat. nº : C0001 (pSpark® I) C0002 (pSpark® II) C0003 (pSpark® III) C0004 (pSpark® IV) C0005 (pSpark® V) C0006 (pSpark® Done) Upon Receipt Store Kits at -20°C PRODUCT MANUAL Version 3.0 Last updated: December 2012 www.canvaxbiotech.com TABLE OF CONTENTS TABLE OF CONTENTS ii MATERIALS PROVIDED, KIT STORAGE AND EXPIRATION DATE iii 1. INTRODUCTION 1 1.1 Principle and advantages 1 1.2 The family of pSpark® DNA cloning vectors 2 1.3 pSpark® DNA cloning vector maps 3 1.4 Specialized applications of the pSpark® DNA cloning systems 4 1.5 Additional materials required (but NOT supplied with kits unless otherwise stated)4 2. DETAILED PROTOCOL 5 2.1 Experimental outline 5 2.2 PCR 6 2.2.1 PCR Primers design 6 2.2.2 PCR Amplification 6 2.3 Ligation 8 2.3.1 Amount of insert needed for ligation into pSpark® DNA cloning systems. 8 2.3.2 Protocol for ligation using the pSpark® DNA cloning systems 10 2.3.3 Tips for cloning of long or problematic PCR products. 11 2.4 Transformation 12 2.4.1 General considerations about transformation into E coli 12 2.4.2 Standard protocol for transformation 13 2.4.3 Fast transformation protocol (Recommended alternative) 14 2.4.4 Transformation by electroporation protocol 16 2.5 Selection of recombinants 17 2.5.1 Expected results 17 2.6 Analysis of transformants 20 2.6.1 PCR directly from bacterial colonies (Colony PCR protocol) 20 2.6.2 Isolation of plasmid DNA 21 2.6.3 Sequencing 22 2.6.4 Long term storage of sequence-verified clones 22 3. -

To Obtain Approval for Projects to Develop Genetically Modified Organisms in Containment
APPLICATION FORM Containment – GMO Project To obtain approval for projects to develop genetically modified organisms in containment Send to Environmental Protection Authority preferably by email ([email protected]) or alternatively by post (Private Bag 63002, Wellington 6140) Payment must accompany final application; see our fees and charges schedule for details. Application Number GMO17LU001 Date 26 June 2017 www.epa.govt.nz 2 Application Form Approval for projects to develop genetically modified organisms in containment Completing this application form 1. This form has been approved under section 42A of the Hazardous Substances and New Organisms (HSNO) Act 1996. It only covers projects for development (production, fermentation or regeneration) of genetically modified organisms in containment. This application form may be used to seek approvals for a range of new organisms, if the organisms are part of a defined project and meet the criteria for low risk modifications. Low risk genetic modification is defined in the HSNO (Low Risk Genetic Modification) Regulations: http://www.legislation.govt.nz/regulation/public/2003/0152/latest/DLM195215.html. 2. If you wish to make an application for another type of approval or for another use (such as an emergency, special emergency or release), a different form will have to be used. All forms are available on our website. 3. It is recommended that you contact an Advisor at the Environmental Protection Authority (EPA) as early in the application process as possible. An Advisor can assist you with any questions you have during the preparation of your application. 4. Unless otherwise indicated, all sections of this form must be completed for the application to be formally received and assessed. -

Gene Cloning
PLNT2530 2021 Unit 6a Gene Cloning Vectors Molecular Biotechnology (Ch 4) Principles of Gene Manipulation (Ch 3 & 4) Analysis of Genes and Genomes (Ch 5) Unless otherwise cited or referenced, all content of this presenataion is licensed under the Creative Commons License 1 Attribution Share-Alike 2.5 Canada Plasmids Gene 1 Naturally occurring plasmids ori -occur widely in bacteria -are covalently closed circular dsDNA -are replicons, stably inherited as extra-chromosomal DNA -can be 1 kbp to 500 kbp in size (compared to 4000 kbp chromosome) -bacteria can contain several different types of plasmid simultaneously -many naturally occurring plasmids carry genes for restriction enzymes, antibiotic resistance, or other genes 2 Bacterial Vectors All vectors : 1. -must replicate autonomously in ori - origin of replication their specific host even when sequence at which DNA polymerase joined to foreign DNA initiates replication 2. - should be easily separated from host chromosomal DNA E. coli chromosomal DNA: ~ 4 million bp typical plasmid vector: ~ 3 to 10 kb Most modern cloning vectors in E. coli are derived from naturally-ocurring plasmid col E1. Most of col E1 was deleted except for an origin of replication and an antibiotic resistance gene. 3 Vectors Types cloning small plasmids- can occur naturally in as circular dsDNA in fragments bacteria (up to 15 kb) eg. single genes bacteriophage -viruses of bacteria (~10-50 kb) used in the cDNA cloning, high-efficiency construction of cDNA and genomic libraries cloning BAC-bacterial artificial chromosome (130-150 kb genomic libraries YAC-Yeast artificial chromosome (1000-2000 kb) with large inserts Each type of vector has specific applications but primary function is to carry foreign DNA (foreign to bacteria) and have it replicated by the bacteria 4 Introduction of foreign DNA into E. -

Lab Exercise: Transformation
Lab Exercise: Transformation OBJECTIVES 1. Understand the process of transformation and how it is used in a laboratory setting for expression of genes (i.e. production of proteins). 2. Perform a successful transformation using the pTOM plasmid. INTRODUCTION Genetic transformation is used in many areas of biotechnology, and, at its heart, requires two things: Donor DNA and recipient cells. Cells which receive the donor DNA are considered genetically recombined, that is, they have their original DNA (their chromosome) and new DNA (the plasmid) and whatever genes that plasmid carries. Before considering the details of recombination, we will consider each of these players individually. Plasmids were discovered as extra-chromosomal genetic material in the late 1960s. Like the bacterial chromosome, they are circular but they are much smaller (2,000–10,000 bp), and usually contain genes for one or more traits that may be beneficial to bacterial survival. In nature, bacteria can transfer plasmids back and forth allowing them to share these beneficial genes and adapt to new environments. For instance, the quick dissemination of bacterial resistance to antibiotics is due in part to the transmission of plasmids. These naturally occurring plasmids have been engineered to contain not only antibiotic resistance (which is used in the laboratory as a selective marker for successful transformation) but other “genes of interest.” If a plasmid is transformed into an E. coli cell that is sensitive to the antibiotic ampicillin, it will confer resistance to that antibiotic. Growing the transformants in the presence of ampicillin is an easy way to select for recombined E. -

A Biobrick Compatible Strategy for Genetic Modification of Plants Boyle Et Al
A BioBrick compatible strategy for genetic modification of plants Boyle et al. Boyle et al. Journal of Biological Engineering 2012, 6:8 http://www.jbioleng.org/content/6/1/8 Boyle et al. Journal of Biological Engineering 2012, 6:8 http://www.jbioleng.org/content/6/1/8 METHODOLOGY Open Access A BioBrick compatible strategy for genetic modification of plants Patrick M Boyle1†, Devin R Burrill1†, Mara C Inniss1†, Christina M Agapakis1,7†, Aaron Deardon2, Jonathan G DeWerd2, Michael A Gedeon2, Jacqueline Y Quinn2, Morgan L Paull2, Anugraha M Raman2, Mark R Theilmann2, Lu Wang2, Julia C Winn2, Oliver Medvedik3, Kurt Schellenberg4, Karmella A Haynes1,8, Alain Viel3, Tamara J Brenner3, George M Church5,6, Jagesh V Shah1* and Pamela A Silver1,5* Abstract Background: Plant biotechnology can be leveraged to produce food, fuel, medicine, and materials. Standardized methods advocated by the synthetic biology community can accelerate the plant design cycle, ultimately making plant engineering more widely accessible to bioengineers who can contribute diverse creative input to the design process. Results: This paper presents work done largely by undergraduate students participating in the 2010 International Genetically Engineered Machines (iGEM) competition. Described here is a framework for engineering the model plant Arabidopsis thaliana with standardized, BioBrick compatible vectors and parts available through the Registry of Standard Biological Parts (www.partsregistry.org). This system was used to engineer a proof-of-concept plant that exogenously expresses the taste-inverting protein miraculin. Conclusions: Our work is intended to encourage future iGEM teams and other synthetic biologists to use plants as a genetic chassis. -

Plasmids 101
Plasmids 101 A Desktop Resource Created and Compiled by Addgene www.addgene.org March 2017 (3rd Edition) Plasmids 101: A Desktop Resource (3rd Edition) INTRODUCTION TO PLASMIDS 101 By The Addgene Team | March, 2017 Any newcomer who joins a molecular biology lab will undoubtedly be asked to design, modify, or construct a plasmid. Although the newcomer likely knows that a plasmid is a small circular piece of DNA often found in bacterial cells, additional guidance may be required to understand the specific components that make up a plasmid and why each is important. Our mission with this eBook, Plasmids 101: A Desktop Resource, is to curate a onestop reference guide for plasmids. This resource is designed to educate all levels of scientists and plasmid lovers. It serves as an introduction to plasmids, allowing you to spend less time researching basic plasmid features and more time developing the clever experiments and innovative solutions necessary for advancing your field. ~ The Addgene Team addgene.org blog.addgene.org facebook.com/addgene twitter.com/addgene linkedin.com/company/addgene youtube.com/addgene blog.addgene.org/topic/podcast 2 | Page Table of Contents Plasmids 101: A Desktop Resource (3rd Edition) TABLE OF CONTENTS Page Section 2 Introduction to Plasmids 101 7 Chapter 1: What is a Plasmid? 8 A Brief History of Plasmids 10 What is a Plasmid? 12 Antibiotic Resistance Genes 14 Common Antibiotics Table 15 Origin of Replication 18 The Promoter Region 25 Terminators and PolyA Signals 28 Methylation and Restriction Enzymes 31 Blue-White Screening 34 Common Lab E. coli Strains 39 E. -

A Cassette with Seven Unique Restriction Sites, Including Octanucleotide Sequences: Extension of Multiple-Cloning-Site Plasmids
Gene, 80 (1989) 151-154 151 Elsevier GENE 03013 A cassette with seven unique restriction sites, including octanucleotide sequences: extension of multiple-cloning-site plasmids (Recombinant DNA; vector; NotI; S’I; oligodeoxyribonucleotides; plasmids) J&g D. Hoheisel Fakultiit ftir Biologie, UniversitiitKonstanz, D-7750 Konstanz (F.R. G.) Received by G. Wilcox: 3 January 1989 Accepted: 27 January 1989 SUMMARY A 27-bp synthetic DNA cassette was constructed which contains the restriction sites of the two rare-cutter enzymes Not1 and S’I and, in an overlapping arrangement, those of live enzymes with 6-bp recognition sequences: &I, Bali, NdeI, SucII, XmaIII. The protruding termini of the fragment allow its insertion into any EcoRI-cut DNA creating a new EcoRI site at one side of the cassette only. This fragment was integrated into the pUC184ke multiple-cloning-site (MCS) plasmids pTZ18R and pTZ 19R, producing a set of vectors which carry seven additional unique restriction sites (giving a total of 17) within their MCS. They still provide the capabilities of simple recombinant selection by blue/white coloured colonies, creation of single-stranded DNA in the presence of a helper phage, and in vitro transcription of cloned DNA using T7 RNA-polymerase. Plasmids with two copies of the DNA cassette inserted into their MCS were also constructed. Beside the advantages they provide in some cloning procedures, these latter plasmids, which carry a tandem repeat, are valuable sources of related 27-bp fragments, with features similar to the original but with different cloning termini. INTRODUCTION tion of such sites in short MCS. That of the plasmid pUC18 (Yanisch-Perron et al., 1985), which pro- Vectors are a fundamental tool in molecular vides a simple selection of recombinant clones due to biology and genetic engineering. -

Cloning Vector Puc19
Cloning Vector pUC19 Product Information Sheet # V33202 SUMMARY shipped at RT; store at 4 °C For research use only Product pUC19 high copy cloning vector for replication in E. coli, suitable for “blue-white screening” technique. Description pUC19 is a small, high copy cloning vector for replication in E. coli. It has been constructed using the ampicillin resistance gene and the pMB1 origin of replication from pBR322. The pMB1 of pUC19 differs from the pBR322 origin by a single point mutation and the lack of the rop gene, leading to a high copy number. Additionally, pUC19 contains the lac operon of E. coli with CAP binding site, lac promoter (Plac), Lac repressor (LacR) binding site, and the 5’-terminal part of the lacZ gene encoding for the N-terminal part of β-galactosidase (source – M13mp19 phage vector). This 5’-terminal part of the lacZ gene contains the multiple cloning site (MCS), and its expression is IPTG inducible. It is capable of intra-allelic α- complementation of a partial deleted chromosomal lacZ copy (E. coli host strain: lacZΔM15, e.g., DH5α, DH10B, JM101, JM109). In the presence of IPTG, transformants expressing both fragments of the ß- galactosidase (the vector encoded N-terminal part and the chromosomal encoded C-terminal part) will form a functional enzyme and can be detected as blue colonies on agar plates containing X-Gal. Cloning into the multiple cloning site will lead to a nonfunctional N-terminal fragment of the ß-galactosidase and to the abolishment of α-complementation. White colonies will form on X-Gal/IPTG plates. -

Designing and Reconstruction of Pcdna 3.0 Mammalian Expression
& Trans Naaz and Kazim, Clon Transgen 2018, 7:1 g ge in n n e s DOI: 10.4172/2168-9849.1000162 o l i s C Cloning & Transgenesis ISSN: 2168-9849 Research Article Open Access Designing and Reconstruction of pcDNA 3.0 Mammalian Expression Vector with its Multiple Cloning Sites by Directional Cloning Method Sheeba Naaz* and Syed Naqui Kazim Centre for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia, New Delhi, India Abstract Traditional cloning is the cloning in which we use restriction endonucleases to produce DNA fragments with specific complementary end sequences that can be joined together with a DNA ligase enzyme, prior to the transformation. The purpose of this study was to reconstruct the mammalian expression vector by replacing the junk DNA with its natural multiple cloning sites. Plasmid was propagated and digested with Xho1 and Kpn1 enzymes from both sides of junk DNA a 600 bp DNA was cut from the vector and then ligation of 30 bp primer which was designed similar as its multiple sequence sites. Correct insertion was confirmed by plasmid isolation, plasmid colony PCR, PCR of cloned plasmid and comparison on gel electrophoresis with the original one and finally by sequencing. An extra DNA fragment of 600 bp was cut from the vector after restriction digestion. Ligated colonies appeared on the agar plate from which 60 bp colony PCR products was produced. Plasmid isolation after cloning and colony PCR and PCR of cloned plasmid confirmed the cloning. Cloning of multiple cloning sites in pcDNA3.0 mammalian expression vector was performed successfully. -
Subcloning Notebook Guide, BR152
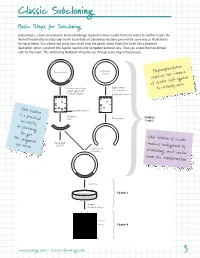
Classic Subcloning Basic Steps for Subcloning Subcloning is a basic procedure in molecular biology required to move inserts from one vector to another to gain the desired functionality to study your insert. Essentially all subcloning reactions proceed the same way as illustrated in the figure below. You release and purify your insert from the parent vector, ligate this insert into a prepared destination vector, transform this ligation reaction into competent bacterial cells. Then you screen the transformed cells for the insert. This Subcloning Notebook will guide you through every step in the process. Destination Parent Vector Vector Dephosphorylation reduces the chance of vector self-ligation Release insert from Digest vector parent vector with to accept insert to virtually zero. restriction digests Dephosphorylation Gel isolation band of interest is a practical Gel-purify Chapters insert Purify vector necessity 1 and 2 in subcloning. You get the fragment Gel isolation of vector Generated you need. by PCR reduces background by Ligate insert to vector eliminating uncut vector from the transformation. Transform Chapter 3 Screen Miniprep Digest 1 2 3 4 –Vector –Insert Chapter 4 4497TA www.promega.com • [email protected] 3 Classic Subcloning Subcloning Strategy See the Compatible Ends Before you begin your subcloning, you need to know: The restriction enzyme Table on page 61 for (RE) sites available for subcloning in your parent vector multiple cloning region (or in the insert if you need to digest the insert); the RE sites available a listing of overhangs in the destination vector multiple cloning region (MCR); and if these same compatible with sites also occur in your insert.