Process for the Preparation of a 2-Substituted-3,3-Difluorofuran
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthesis of Protected 2′-Deoxy-2′-Fluoro-Β-D-Arabinonucleosides
Synthesis of Protected UNIT 1.7 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides This unit describes in detail the preparation of protected 2′-deoxy-2′-fluoroarabinonu- cleosides. These building blocks are required for the synthesis of 2′-deoxy-2′-fluoroarabi- nonucleic acid (2′F-ANA), an oligonucleotide analog exhibiting very promising antisense properties (Damha et al., 1998; Wilds and Damha, 2000; Lok et al., 2002). The preparation of phosphoramidites from these building blocks and the synthesis of 2′F-ANA are described in UNIT 4.15. SYNTHESIS AND CHARACTERIZATION OF N2-ISOBUTYRYL-9- BASIC [2-DEOXY-2-FLUORO-5-O-(4-METHOXYTRITYL)-β-D- PROTOCOL 1 ARABINOFURANOSYL]GUANINE Synthesis of araF-G (S.6; Figure 1.7.3) was accomplished via the condensation of 2,6-dichloropurine with either 2-deoxy-2-fluoro-1,3,5-tri-O-benzoyl-α-D-arabinofura- nose (S.2; Figure 1.7.1) or 2-deoxy-2-fluoro-3,5-di-O-benzoyl-α-D-arabinofuranosyl bromide (S.3) as a key chemical step (Figure 1.7.2). Starting from S.3 gives a higher yield of S.4 and simplifies its purification. The versatile intermediate N9-β-glycoside (S.4) was transformed into araF-G in three steps (Figure 1.7.3). Treatment of S.4 with NaN3 in ethanol gave the 2,6-diazido derivative (Wower et al., 1994). This product was then subjected to reduction with SnCl2 in a mixture of dichloromethane/methanol to give its 2,6-diamino derivative (Tennila et al., 2000). Standard debenzoylation and deamination gave araF-G. -

Carbon–Fluorine Bond Formation
Carbon–Fluorine Bond Formation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Furuya, Takeru, Christian A. Kuttruff, and Tobias Ritter. 2008. Carbon–fluorine bond formation. Current Opinion in Drug Discovery and Development 11(6): 803–819. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:8301597 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Open Access Policy Articles, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#OAP Carbon–Fluorine Bond Formation Takeru Furuya, Christian A. Kuttruff, and Tobias Ritter* Department of Chemistry and Chemical Biology Harvard University 12 Oxford Street Cambridge, MA 02138 [email protected] Phone 617 496 0750 Fax 617 496 4591 1 Abstract We present a selection of carbon–fluorine bond formations that have been developed in the recent past. An overview of the most common fluorination reagents is followed by fluorination reactions organized by reactivity. We have distinguished between nucleophilic and electrophilic fluorinations as well as aliphatic and aromatic fluorinations. Each section is divided into more specific reaction classes and examples for syntheses of pharmaceuticals, 18F-radiolabeling, and mechanistic investigations are provided. Keywords Fluorination; carbon–fluorine bond formation; nucleophilic fluorination; electrophilic fluorination; fluorinating -
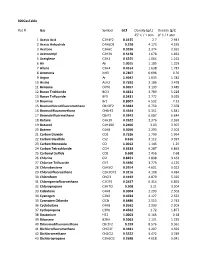
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Oxidation Trifluoride (DAST) Selectfluor™
Vol. 5 No. 1 Synthetic Methods Iodotoluene difluoride Diethylaminosulfur Oxidation trifluoride (DAST) Selectfluor™ Ishikawa’s Reagent Other Reagents for Fluorination Dess–Martin Periodinane Bis(pyridine)iodonium tetrafluoroborate Magnesium bis(monoperoxyphthalate) hexahydrate Chloramine-T Other Reagents for Oxidation Advancing Science 2 Introduction Oxidation is one of the most common transformations in all of organic chemistry. As a result, the number of oxidizing agents and reactions at the research chemist’s disposal number in the hundreds; Sigma-Aldrich is proud to offer a new series of ChemFiles—called however, few provide extended utility across a broad range of Synthetic Methods—to our Organic Chemistry and Drug Discovery substrates while remaining able to selectively oxidize one group customers. Each piece will highlight a specific motif or selected over another. Fewer yet remain effective under mild conditions. At organic transformation, and the range of products Sigma-Aldrich Sigma-Aldrich, we are committed to being your preferred supplier offers in this area. Our first installment focuses on oxidation for reagents used in oxidation—and we’d like to highlight some chemistry. In the future, we will highlight other synthetic methods new, popular, and versatile oxidation reagents in this ChemFile. such as asymmetric synthesis and reduction. We trust you will find these pieces useful, and we welcome your comments for Listed on the following pages is a selection of oxidation reagents future installments. If you cannot find a product for your specific available from Sigma-Aldrich. For a complete listing of oxidation research in organic synthesis or drug discovery, we encourage your reagents, please visit sigma-aldrich.com/oxidation. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Dialkylaminodifluorosulfinium Tetrafluoroborate Salts
Process optimization Dialkylaminodifluorosulfinium Marc-Olivier Turcotte-Savard tetrafluoroborate salts: synthesis and applications MARC-OLIVIER TURCOTTE-SAVARD1*, OLIVIER MAHÉ2, JEAN-FRANÇOIS PAQUIN2 *Corresponding author 1. OmegaChem, 480, Rue Perreault, Lévis, QC, G6W 7V6, Canada 2. Université Laval, Département de chimie, Canada Research Chair in Organic and Medicinal Chemistry, CCVC, PROTEO, 1045 avenue de la Médecine, Québec, QC, G1V 0A6, Canada various fluoride ion acceptors (PF , SeF , SbF and AsF ) along KEyWORDS 5 4 5 5 with DAST-type reagents (7 - 10). However, the reactivity of these Organofluorine chemistry; deoxyfluorination; salts has been scarcely studied until 2009. C-F bond; XtalFluor; dialkylaminodifluorosulfinium Recently, a one-pot preparation of diethylaminodifluorosulfinium tetrafluoroborate salts tetrafluoroborate has been developed. Hence, the addition of THF•BF3 adduct to crude DAST, previously formed from SF4 in ABSTRACT CH2Cl2 and N,N-diethyltrimethylsilylamine, resulted in 90% yield of diethylaminodifluorosulfinium tetrafluoroborate 1 (4). The solid The syntheses of dialkylaminodifluorosulfinium obtained was under a different polymorphic form with higher tetrafluoroborate salts are described. These reagents, melting point (89.8°C vs. 74-76°C) and lower moisture sensitivity first used for the deoxyfluorination of alcohols, compared to the first polymorph reported by Markovskii (5). were also shown to fluorinate aldehydes, ketones, carboxylic acids and glycosyl groups. Alternatively, dialkylaminodifluorosulfinium tetrafluoroborate salts were 14 recently shown to act as activating agents for hydroxyl, carbonyl and carboxyl groups in a variety of transformations leading to non-fluorinated products. Scheme 1. INTRODUCTION Similarly, addition of boron trifluoride etherate to an ice cold solution of crude morpholinosulfur trifluoride gave an excellent ith 30 to 40% of agrochemicals and 20 to 25% of yield of the corresponding tetrafluoroborate salt 2 (4). -

United States Patent (19) 11 4,311,651 Middleton 45) Jan
United States Patent (19) 11 4,311,651 Middleton 45) Jan. 19, 1982 54 FLUOROALKOXYSULFUR FLUORIDES Darragh J. I. et al., Angew. Chem. Int. 9, 73 (1970). 75) Inventor: William J. Middleton, Chadds Ford, Middleton, W. J., J. Org. Chem. 40, No. 5 pp. 574-578 Pa. (1975). w 73) Assignee: E. I. Du Pont de Nemours and Primary Examiner-Paul J. Killos Company, Wilmington, Del. Attorney, Agent, or Firm-James A. Costello 21 Appl. No.: 148,209 57 ABSTRACT 22 Filed: May 9, 1980 Fluoroalkoxysulfur fluorides of the formula 51) Int. Cl. ................... C07C 143/00; CO7C 147/00 52 U.S. C. ................................. 260/543 H; 568/27; 568/842; 568/939; 568/940; 260/239 BD f 58) Field of Search ...................... 260/543 H; 568/27 R--o (56) References Cited CF U.S. PATENT DOCUMENTS wherein R is H, CH3, CF3, or C2F5, and n is 1 or 2; their 3,418,337 12/1968 Middleton ........................ 260/347.8 preparation by reaction of lithium fluoroalkoxides with 3,888,924 6/1975 Middleton ... 260/543 F sulfur tetrafluoride; their use as fluorinating agents to 3,914,265 10/1975 Middleton ........................ 260/397.3 replace OH groups in organic molecules with F; and OTHER PUBLICATIONS halocarbon solutions of lithium fluoroalkoxides. Baum, K, J.A.C.S. 914594 (1969). Darragh, J. I. et al., J.C.S. Dalton Trans., 1975, 218. 13 Claims, No Drawings 4,311,651 1. 2 wherein R is as defined above. FLUOROALKOXYSULFUR FLUORIDES The fluoroalkoxysulfur fluorides of this invention are useful as fluorinating agents. In particular, they are BACKGROUND OF THE INVENTION useful for replacing hydroxyl groups of organic com 1. -

Summer Assignment 2018 the Summer Assignment Is to Help
Summer Assignment 2018 The summer assignment is to help prepare you for the beginning of AP chemistry. The assignment is not graded, but the test on the second day of school covering the material will be the summer assignment grade. The packet contains the following: 1. My general chemistry notes (Most of the pages ) 2. Element and polyatomic ion lists 3. Practice problems For the test, you must know the following: 1. Have all the common elements memorized – name and symbol 2. Have ALL the polyatomic ions memorized 3. Be able to write from symbol to word and word to symbol covalent and ionic compounds. 4. Be able to balance an equation 5. Memorize the diatomic molecules The test will have the following: 1. 10 equations where the products and reactants will be in word form. You will write the symbol and balance the reaction. Example: Sodium chloride and magnesium oxide make sodium oxide and magnesium chloride. Answer: 2NaCl + MgO Na2O + MgCl2 I will be having a study session June 20th – the last day of school. It is not required but it will be a good introduction to what you need to know for the summer assignment test. I would recommend attending; it takes an hour at the most. I will be sending out a letter in early June with the time of the session. I will be having a study session in August to help with the summer assignment. The date have not been determined and will not be until August. PLEASE EMAIL ME YOUR EMAIL ADDRESS AT THE BEGINNING OF THE SUMMER SO I CAN EMAIL YOU THE DAYS AND TIMES OF THE STUDY SESSIONS! The session are not required, but helpful. -
![United States Patent 19 [11] 3,940,402 Middleton (45) Feb](https://docslib.b-cdn.net/cover/1860/united-states-patent-19-11-3-940-402-middleton-45-feb-4081860.webp)
United States Patent 19 [11] 3,940,402 Middleton (45) Feb
United States Patent 19 [11] 3,940,402 Middleton (45) Feb. 24, 1976 54 TRIS(SUBSTITUTED AMINO) SULFONIUM OTHER PUBLICATIONS SALTS (75) Inventor: William Joseph Middleton, Chadds J.A.C.S. 84:3374-3387, (1962), Melby et al. Ford, Pa. 73 Assignee: E. I. DuPont de Nemours and Primary Examiner-Sherman D. Winters Company, Wilmington, Del. Attorney, Agent, or Firm-Anthony P. Mentis 22) Filed: Sept. 19, 1974 21) Appl. No.: 507,426 57 ABSTRACT Trisaminosulfonium salts of the formula 52 U.S. C. ...... 260/293.63; 260/80 C; 260/239 B; (RRN)(R'R''N)(R5RN)St X wherein the R groups 260/293.51; 260/293.85; 260/326.61; are alkyl of 1-20 carbons and each alkyl has at least 2 260/326.62; 260/326.82; 260/583 R; alpha hydrogens and X is selected from the group 260/583 EE (CH)SiF, Cl, Br, I, CN, NCO, NCS NO, and N, are 5 Int. Cl.'........................................ C07D 295/22 soluble in organic liquids. They are useful as polymeri 58) Field of Search..... 260/293.63, 326.61, 326.62, zation catalysts and as reagents to replace other atoms 260/326.82, 583 EE, 293.85 or groups in organic compounds with F, Cl, Br, I, CN, NCO, NCS, NO, O N. 56 References Cited UNITED STATES PATENTS 28 Claims, No Drawings 3,162,641 12/1964 Acker et al......................... 260/286 3,940,402 1. 2 The triaminosulfonium difluorotrimethylsilicates of TRES(SUBSTITUTED AMINO) SULFONIUM SALTS the invention are prepared by treating a solution of sulfur tetrafluoride in an anhydrous inert solvent with BACKGROUND OF THE INVENTION at least three equivalents of a (secondary amino)trime 1. -

Enthalpies of Vaporization of Organic and Organometallic Compounds, 1880–2002
Enthalpies of Vaporization of Organic and Organometallic Compounds, 1880–2002 James S. Chickosa… Department of Chemistry, University of Missouri-St. Louis, St. Louis, Missouri 63121 William E. Acree, Jr.b… Department of Chemistry, University of North Texas, Denton, Texas 76203 ͑Received 17 June 2002; accepted 17 October 2002; published 21 April 2003͒ A compendium of vaporization enthalpies published within the period 1910–2002 is reported. A brief review of temperature adjustments of vaporization enthalpies from temperature of measurement to the standard reference temperature, 298.15 K, is included as are recently suggested reference materials. Vaporization enthalpies are included for organic, organo-metallic, and a few inorganic compounds. This compendium is the third in a series focusing on phase change enthalpies. Previous compendia focused on fusion and sublimation enthalpies. Sufficient data are presently available for many compounds that thermodynamic cycles can be constructed to evaluate the reliability of the measure- ments. A protocol for doing so is described. © 2003 American Institute of Physics. ͓DOI: 10.1063/1.1529214͔ Key words: compendium; enthalpies of condensation; evaporation; organic compounds; vaporization enthalpy. Contents inorganic compounds, 1880–2002. ............. 820 1. Introduction................................ 519 8. References to Tables 6 and 7.................. 853 2. Reference Materials for Vaporization Enthalpy Measurements.............................. 520 List of Figures 3. Heat Capacity Adjustments. ................. 520 1. A thermodynamic cycle for adjusting vaporization ϭ 4. Group Additivity Values for C (298.15 K) enthalpies to T 298.15 K.................... 521 pl 2. A hypothetical molecule illustrating the different Estimations................................ 521 hydrocarbon groups in estimating C ........... 523 5. A Thermochemical Cycle: Sublimation, p Vaporization, and Fusion Enthalpies........... -

Exploring the Interaction Between Asymmetric Phosphonates and Acetylcholinesterase. Probing the Gorge and P-Site Via Customized Covalent Modification
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 2008 Exploring the interaction between asymmetric phosphonates and acetylcholinesterase. Probing the gorge and P-site via customized covalent modification Lilu Guo The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Guo, Lilu, "Exploring the interaction between asymmetric phosphonates and acetylcholinesterase. Probing the gorge and P-site via customized covalent modification" (2008). Graduate Student Theses, Dissertations, & Professional Papers. 421. https://scholarworks.umt.edu/etd/421 This Dissertation is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. EXPLORING THE INTERACTION BETWEEN ASYMMETRIC PHOSPHONATES AND ACETYLCHOLINESTERASE. PROBING THE GORGE AND P-SITE VIA CUSTOMIZED COVALENT MODIFICATION. By Lilu Guo B.S., Peking University, Beijing, P.R.China, 2000 Dissertation presented in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry The University of Montana Missoula, MT Summer 2008 Approved by: Dr. Perry Brown, Associate Provost Graduate School Dr. Charles M. Thompson, Chair Department of Biomedical & Pharmaceutical Sciences Dr. Mark Cracolice Department of Chemistry Dr. C. Sean Esslinger Department of Biomedical & Pharmaceutical Sciences Dr. J.B. Alexander (Sandy) Ross Department of Chemistry Dr. Holly Thompson Department of Chemistry © COPYRIGHT by Lilu Guo 2008 All Rights Reserved Guo, Lilu, PhD., Summer 2008 Chemistry Exploring the interaction between asymmetric phosphonates and acetylcholinesterase. -

Presidential Green Chemistry Challenge Awards Program: Summary of 2000 Award Entries and Recipients
United States Pollution Prevention and EPA744-R-00-001 Environmental Protection Toxics (7406) August 2001 Agency www.epa.gov/greenchemistry 1EPA The Presidential Green Chemistry Challenge Awards Program Summary of 2000 Award Entries and Recipients 2 Printed on paper that contains at least 50% postconsumer fiber. The Presidential Green Chemistry Challenge Awards Program Contents Summary of 2000 Award Entries and Recipients . 1 Awards . 3 Academic Award . 3 Small Business Award . 4 Alternative Synthetic Pathways Award . 5 Alternative Solvents/Reaction Conditions Award . 6 Designing Safer Chemicals Award . 7 Entries From Academia . 9 Entries From Small Businesses . 21 Entries From Industry and Government . 33 Index . 55 i The Presidential Green Chemistry Challenge Awards Program Summary of 2000 Award Entries and Recipients The Pollution Prevention Act of 1990 established a national policy to prevent or reduce pollution at its source whenever feasible. Green chemistry, the design of chemical products and processes that reduce or eliminate the use and generation of hazardous substances, is a highly effective approach to pollution prevention because it applies innovative scientific solu tions to real-world environmental situations, all through voluntary partnership programs. The Presidential Green Chemistry Challenge promotes pollution prevention and indus trial ecology through an EPA (U.S. Environmental Protection Agency) Design for the Environment partnership with the chemistry community. Through high level recognition and support, the Presidential Green Chemistry Challenge promotes innovative developments in and uses of green chemistry for pollution prevention. The technologies recognized and supported by the Presidential Green Chemistry Challenge directly reduce risks to human health and the environment by reducing the hazards associated with the design, manufacture, and use of chemicals.