Preparation of Phenylsulphur Chloride Tetrafluoride
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthesis of Protected 2′-Deoxy-2′-Fluoro-Β-D-Arabinonucleosides
Synthesis of Protected UNIT 1.7 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides This unit describes in detail the preparation of protected 2′-deoxy-2′-fluoroarabinonu- cleosides. These building blocks are required for the synthesis of 2′-deoxy-2′-fluoroarabi- nonucleic acid (2′F-ANA), an oligonucleotide analog exhibiting very promising antisense properties (Damha et al., 1998; Wilds and Damha, 2000; Lok et al., 2002). The preparation of phosphoramidites from these building blocks and the synthesis of 2′F-ANA are described in UNIT 4.15. SYNTHESIS AND CHARACTERIZATION OF N2-ISOBUTYRYL-9- BASIC [2-DEOXY-2-FLUORO-5-O-(4-METHOXYTRITYL)-β-D- PROTOCOL 1 ARABINOFURANOSYL]GUANINE Synthesis of araF-G (S.6; Figure 1.7.3) was accomplished via the condensation of 2,6-dichloropurine with either 2-deoxy-2-fluoro-1,3,5-tri-O-benzoyl-α-D-arabinofura- nose (S.2; Figure 1.7.1) or 2-deoxy-2-fluoro-3,5-di-O-benzoyl-α-D-arabinofuranosyl bromide (S.3) as a key chemical step (Figure 1.7.2). Starting from S.3 gives a higher yield of S.4 and simplifies its purification. The versatile intermediate N9-β-glycoside (S.4) was transformed into araF-G in three steps (Figure 1.7.3). Treatment of S.4 with NaN3 in ethanol gave the 2,6-diazido derivative (Wower et al., 1994). This product was then subjected to reduction with SnCl2 in a mixture of dichloromethane/methanol to give its 2,6-diamino derivative (Tennila et al., 2000). Standard debenzoylation and deamination gave araF-G. -

Carbon–Fluorine Bond Formation
Carbon–Fluorine Bond Formation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Furuya, Takeru, Christian A. Kuttruff, and Tobias Ritter. 2008. Carbon–fluorine bond formation. Current Opinion in Drug Discovery and Development 11(6): 803–819. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:8301597 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Open Access Policy Articles, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#OAP Carbon–Fluorine Bond Formation Takeru Furuya, Christian A. Kuttruff, and Tobias Ritter* Department of Chemistry and Chemical Biology Harvard University 12 Oxford Street Cambridge, MA 02138 [email protected] Phone 617 496 0750 Fax 617 496 4591 1 Abstract We present a selection of carbon–fluorine bond formations that have been developed in the recent past. An overview of the most common fluorination reagents is followed by fluorination reactions organized by reactivity. We have distinguished between nucleophilic and electrophilic fluorinations as well as aliphatic and aromatic fluorinations. Each section is divided into more specific reaction classes and examples for syntheses of pharmaceuticals, 18F-radiolabeling, and mechanistic investigations are provided. Keywords Fluorination; carbon–fluorine bond formation; nucleophilic fluorination; electrophilic fluorination; fluorinating -
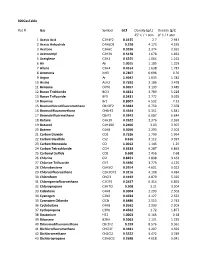
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Oxidation Trifluoride (DAST) Selectfluor™
Vol. 5 No. 1 Synthetic Methods Iodotoluene difluoride Diethylaminosulfur Oxidation trifluoride (DAST) Selectfluor™ Ishikawa’s Reagent Other Reagents for Fluorination Dess–Martin Periodinane Bis(pyridine)iodonium tetrafluoroborate Magnesium bis(monoperoxyphthalate) hexahydrate Chloramine-T Other Reagents for Oxidation Advancing Science 2 Introduction Oxidation is one of the most common transformations in all of organic chemistry. As a result, the number of oxidizing agents and reactions at the research chemist’s disposal number in the hundreds; Sigma-Aldrich is proud to offer a new series of ChemFiles—called however, few provide extended utility across a broad range of Synthetic Methods—to our Organic Chemistry and Drug Discovery substrates while remaining able to selectively oxidize one group customers. Each piece will highlight a specific motif or selected over another. Fewer yet remain effective under mild conditions. At organic transformation, and the range of products Sigma-Aldrich Sigma-Aldrich, we are committed to being your preferred supplier offers in this area. Our first installment focuses on oxidation for reagents used in oxidation—and we’d like to highlight some chemistry. In the future, we will highlight other synthetic methods new, popular, and versatile oxidation reagents in this ChemFile. such as asymmetric synthesis and reduction. We trust you will find these pieces useful, and we welcome your comments for Listed on the following pages is a selection of oxidation reagents future installments. If you cannot find a product for your specific available from Sigma-Aldrich. For a complete listing of oxidation research in organic synthesis or drug discovery, we encourage your reagents, please visit sigma-aldrich.com/oxidation. -

The Synthesis and Characterization of Novel Pentafluorosulfanyl-Containing Heterocycles and Pentafluorosulfanyldifluoromethane
Clemson University TigerPrints All Dissertations Dissertations May 2019 The yS nthesis and Characterization of Novel Pentafluorosulfanyl-Containing Heterocycles and Pentafluorosulfanyldifluoromethane Steven Paul Belina Clemson University, [email protected] Follow this and additional works at: https://tigerprints.clemson.edu/all_dissertations Recommended Citation Belina, Steven Paul, "The yS nthesis and Characterization of Novel Pentafluorosulfanyl-Containing Heterocycles and Pentafluorosulfanyldifluoromethane" (2019). All Dissertations. 2373. https://tigerprints.clemson.edu/all_dissertations/2373 This Dissertation is brought to you for free and open access by the Dissertations at TigerPrints. It has been accepted for inclusion in All Dissertations by an authorized administrator of TigerPrints. For more information, please contact [email protected]. THE SYNTHESIS AND CHARACTERIZATION OF NOVEL PENTAFLUOROSULFANYL-CONTAINING HETEROCYCLES AND PENTAFLUOROSULFANYLDIFLUOROMETHANE A Dissertation Presented to the Graduate School of Clemson University In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Chemistry by Steven Paul Belina May 2019 Accepted by Joseph S. Thrasher, Ph.D., Committee Chair Julia L. Brumaghim, Ph.D. R. Karl Dieter, Ph.D. Joseph W. Kolis, Ph.D. Title Page ABSTRACT Beginning in the early 1960s, scientists began to experiment with the pentafluorosulfanyl moiety. The unique properties of the functional group has attracted interest among fluorine chemists and more recently even organic chemists. These properties include high group electronegativity, high steric bulk, high lipophilicity, a highly electron withdrawing nature, and a square pyramidal geometry. However, the development and deployment of the pentafluorosulfanyl functional group has been significantly slowed. The main cause for the slow development is a lack of easily available reagents to synthesize pentafluorosulfanyl-containing compounds. Historically, the primary reagents used for synthesizing pentafluorosulfanyl-containing compounds were SF5Cl and SF5Br. -

United States Patent As Fice Patiented Sept
s 2,904,403 United States Patent As fice Patiented Sept. 15, 1959 1 2 are agitated during this period by any suitable means. The vessel is then cooled, opened and iodine penta 2,904,403 fluoride collected and purified by conventional procedures such as distillation. When alkali or alkaline earth metal PREPARATION OF BF iodates are used in the process, the iodine pentafluoride William Channing Smith, Wilmington, Del, assignor to may be isolated in the form of a solid complex compound E. H. du Pont de Nemours and Company, Wilmington, with alkali or alkaline earth metal fluorides. Dei, a corporation of Delaware in a continuous process gaseous sulfur tetrafluoride is No Drawing. Application June 7, 1957 passed through a tube containing the iodine compound, Serial No. 664,162 0 preferably heated to a temperature high enough to vola tilize the iodine pentafluoride as it is formed. The 5 Claims. (C. 23-205) iodine pentafluoride is separated from the gas effluent by cooling in traps and the unreacted sulfur tetrafluoride is recycled through the reaction tube. This invention relates to iodine fluorides and has as 5 The following examples illustrate the process of the its primary object provision of a novel method of prepar invention. ing such fluorides, iodine pentafluoride in particular. Iodine pentafluoride is a versatile and well-known Example 1 fluorinating agent which can be used to prepare fluorine (A) A bomb, lined with stainless steel and of 145 mi. bearing compounds that are not readily accessible by 20 capacity, was charged with 33.4 g. (0.10 mode) of iodine other routes. -

United States Patent Office Patented Mar
3,373,000 United States Patent Office Patented Mar. 2, 1968 2 3,373,000 monofluoride is also an economical reactant, since it is PROCESS FOR PREPARING TETRAFLUORDES conveniently prepared by the disproportionation of ele AND HEXAFLUOR DES mental chlorine in liquid hydrogen fluoride, thereby avoid James J. Pitts, West Haven, and Albert W. Jache, North ing the use of expensive elemental fluorine. The selective Haven, Conn., assignors to Olin Mathieson Chemical fluorination process of this invention is particularly Sur Corporation, a corporation of Virginia prising and unexpected in view of the prior art which No Drawing. Filed Nov. 1, 1966, Ser. No. 591,079 teaches that chlorine monofluoride adds to a wide variety 9 Claims. (Cl. 23-352) of compounds, thereby acting as a chloro-fluorinating agent. For instance, U.S. Patent 3,035,893 discloses the 10 preparation of sulfur chloride pentafluoride from Sulfur ABSTRACT OF THE DISCLOSURE tetrafluoride and chlorine monofluoride. The tetrafluorides and hexafluorides of sulfur, selenium According to the process of this invention, the tetra and tellurium are provided by reacting chlorine mono fluorides and hexafluorides of sulfur, selenium and tel fluoride with sulfur, selenium, tellurium, metal sulfides, lurium are provided by reacting chlorine monofluoride metal selenides or metal tellurides. The hexafluorides of 15 with a material selected from the group consisting of Sul tungsten, molybdenum and uranium are provided by the fur, selenium, tellurium, metal sulfides, metal selenides reaction of chlorine monofluoride with the metals, their and metal tellurides. The hexafluorides of tungsten, oxides, sulfides and salts containing the metallate anion. molybdenum and uranium are provided by reacting These tetrafluorides and hexafluorides are well-known chlorine monofluoride with a material selected from the compounds, useful as fluorinating agents, gaseous di 20 group consisting of tungsten; molybdenum, uranium; the electrics, sources of high purity metallic powders, etc. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Sulfur Dioxide and “Sulfurous Acid” Solutions. S + O 2 → SO2 (G
Sulfur Dioxide and “Sulfurous Acid” Solutions. S + O2 → SO2 (g) Structure: sp2 at sulfur, two used for σ bonds, one for lone pair. : δ+ S - δ- δ O O Liquid SO2: Easy to get (bp = -10 °C) and easy to keep liquid at room T under slight pressure. It is quite a good solvent; SO2 is a polar molecule, and the liquid is associated by dipole-dipole attraction. Dissolves polar molecules and ionic compounds, and easy to remove by evaporation. Aqueous solutions: SO2 is soluble in water. Most of it stays in the form of hydrogen bonded “hydrate” (see CO2). At equilibrium in neutral water (no added base) a small portion of it reacts: - H+ - H+ - 2- SO2 (aq) + H2O HO-SO2 SO3 (sulfite - pyramidal) + H+ + H+ - H-SO3 (bisulfite) - 2- 2- Also, 2 HSO3 S2O5 + H2O (structure = [O2S-SO3] ) Many salts of all these anions are known. Complexes formed with sulfite ion and SO2 itself. Sulfur Trioxide and Sulfuric Acid. 2 SO2 + O2 → 2 SO3 (g) Condenses to a liquid at room T, liquid contains monomer and trimer. O O S O O O O O S S S O O O O O Also get solid polymers. SO3 is a powerful oxidant, with water, gives sulfuric acid – H2SO4. A viscous, hydrogen bonded liquid; which is a good strongly acidic solvent. In dilute solutions in water it is a dibasic acid. - H+ - H+ - 2- H2SO4 (aq) HSO4 (bisulfate) SO4 (sulfate) + H+ + H+ Many salts known for both anions. Sulfate can be monodentate or bidentate ligand. SO3 dissolves in concentrated H2SO4 to give “fuming” sulfuric acid which contains some pyrosulfuric acid – VERY CORROSIVE H2SO4 + SO3 H2S2O7 Halides of Sulfur Sulfur hexafluoride (SF6) is a gas a stp. -

Theoretical Bonding Description of Alkylidene Chalcogen (O, S, Se) Difluorides
Inorg. Chem. 1999, 38, 6257-6260 6257 Theoretical Bonding Description of Alkylidene Chalcogen (O, S, Se) Difluorides (H2CdXF2): Planar versus Bent Conformations J. A. Dobado, Henar Martı´nez-Garcı´a, and Jose´ Molina Molina* Grupo de Modelizacio´n y Disen˜o Molecular, Instituto de Biotecnologı´a, Campus Fuentenueva, Universidad de Granada, 18071-Granada, Spain ReceiVed June 23, 1999 Theoretical descriptions of alkylidene chalcogen difluorides have been performed on their planar (1-3) and bent (4-6) conformations. The planar T-shaped conformations were the most stable ones, from the calculations performed (Gaussian-G2 and B3LYP/6-311+G*). The bonding nature of the T-shaped (C2V) structures has also been analyzed by means of the atoms in molecules (AIM) theory and electron localization function analyses. Introduction HF calculations warrant further investigation on the potential The bonding nature of pnicogen and chalcogen ylides has energy surface for these structures, including a detailed descrip- been controversial for years, and recently Gilheany1 has tion of the bonding nature. This theoretical bonding description reviewed the chemical bonding in phosphine ylides. In this should include the electron correlation effects due to the nature - - context, our laboratory has performed calculations on pnicogen2 of the F S F moiety. and chalcogen3 oxides and sulfides, and very recently a general In this paper we present theoretical evidence for the stability study on pnicogen and chalcogen ylides has been presented.4 of planar T-shaped (C2V) alkylidene chalcogen difluorides Alkylidene chalcogen difluorides present special bonding together with the topology characterization of the electron charge F properties, and the sulfur derivatives were first synthesized by density, (r), and the electron localization function (ELF). -

Exemption for the Use of Cadmium in Portable Batteries and Accumulators
Exemption for the use of cadmium in portable batteries and accumulators intended for the use in cordless power tools in the context of the Batteries Directive 2006/66/EC Final Report (revised) 26 January 2010 Consortium ESWI Expert TeamBIPRO, to S upportUmweltbundesamt Waste Implementation BiPRO Beratungsgesellschaft für integrierte Problemlösungen ENV.G.4/FRA/2007/0066 iii European Commission ESWI Final Report - Replacement of Cadmium Batteries in Cordless Power Tools ENV.G.4/FRA/2007/0066 iv Executive Summary Background The Batteries Directive 2006/66/EC (repealing Directive 91/157/EEC) entered into force on 26 September 20061. The Directive sets out rules applicable to all batteries and accumulators that are put on the European Union market. These rules include, among others, restriction on the use of cadmium in portable batteries and accumulators (PBA) according to Article 4(1)(b) of the Directive. Portable batteries and accumulators, including those incorporated into appliances, that contain more than 0,002% of cadmium by weight shall not be placed on the market. However, according to Article 4(3) of the Directive the above requirement shall not apply to portable batteries and accumulators intended for use in: (a) emergency and alarm systems, including emergency lighting; (b) medical equipment; or (c) cordless power tools. Furthermore, based on Article 4(4) of the Directive the Commission shall review the exemption from the cadmium ban for use in cordless power tool with a view to the prohibition of cadmium in batteries and accumulators. The Commission shall submit a corresponding report to the European Parliament and to the Council by 26 September 2010, together, if appropriate, with relevant proposals. -

TIH/PIH List
Hazardous Materials Designated as TIH/PIH (consolidated AAR and Railinc lists) 3/12/2007 STCC Proper Shipping Name 4921402 2-CHLOROETHANAL 4921495 2-METHYL-2-HEPTANETHIOL 4921741 3,5-DICHLORO-2,4,6-TRIFLUOROPYRIDINE 4921401 ACETONE CYANOHYDRIN, STABILIZED 4927007 ACROLEIN, STABILIZED 4921019 ALLYL ALCOHOL 4923113 ALLYL CHLOROFORMATE 4921004 ALLYLAMINE 4904211 AMMONIA SOLUTION 4920360 AMMONIA SOLUTIONS 4904209 AMMONIA, ANHYDROUS 4904210 AMMONIA, ANHYDROUS 4904879 AMMONIA, ANHYDROUS 4920359 AMMONIA, ANHYDROUS 4923209 ARSENIC TRICHLORIDE 4920135 ARSINE 4932010 BORON TRIBROMIDE 4920349 BORON TRICHLORIDE 4920522 BORON TRIFLUORIDE 4936110 BROMINE 4920715 BROMINE CHLORIDE 4918505 BROMINE PENTAFLUORIDE 4936106 BROMINE SOLUTIONS 4918507 BROMINE TRIFLUORIDE 4921727 BROMOACETONE 4920343 CARBON MONOXIDE AND HYDROGEN MIXTURE, COMPRESSED 4920399 CARBON MONOXIDE, COMPRESSED 4920511 CARBON MONOXIDE, REFRIGERATED LIQUID 4920559 CARBONYL FLUORIDE 4920351 CARBONYL SULFIDE 4920523 CHLORINE 4920189 CHLORINE PENTAFLUORIDE 4920352 CHLORINE TRIFLUORIDE 4921558 CHLOROACETONE, STABILIZED 4921009 CHLOROACETONITRILE 4923117 CHLOROACETYL CHLORIDE 4921414 CHLOROPICRIN 4920516 CHLOROPICRIN AND METHYL BROMIDE MIXTURES 4920547 CHLOROPICRIN AND METHYL BROMIDE MIXTURES 4920392 CHLOROPICRIN AND METHYL CHLORIDE MIXTURES 4921746 CHLOROPIVALOYL CHLORIDE 4930204 CHLOROSULFONIC ACID 4920527 COAL GAS, COMPRESSED 4920102 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920303 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920304 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920305 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920101 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920300 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920301 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920324 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920331 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920165 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. 4920378 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. 4920379 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S.