Exploring the Interaction Between Asymmetric Phosphonates and Acetylcholinesterase. Probing the Gorge and P-Site Via Customized Covalent Modification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthetic Routes to Bromo-Terminated Phosphonate Films and Alkynyl Pyridine Compounds for Click Coupling
University of Mary Washington Eagle Scholar Student Research Submissions Spring 5-7-2018 Synthetic Routes to Bromo-Terminated Phosphonate Films and Alkynyl Pyridine Compounds for Click Coupling Poornima Sunder Follow this and additional works at: https://scholar.umw.edu/student_research Part of the Biochemistry Commons Recommended Citation Sunder, Poornima, "Synthetic Routes to Bromo-Terminated Phosphonate Films and Alkynyl Pyridine Compounds for Click Coupling" (2018). Student Research Submissions. 226. https://scholar.umw.edu/student_research/226 This Honors Project is brought to you for free and open access by Eagle Scholar. It has been accepted for inclusion in Student Research Submissions by an authorized administrator of Eagle Scholar. For more information, please contact [email protected]. Synthetic Routes to Bromo-Terminated Phosphonate Films and Alkynyl Pyridine Compounds for Click Coupling Poornima Rachel Sunder Thesis submitted to the faculty of University of Mary Washington in partial fulfillment of the requirements for graduation with Honors in Chemistry (2018) ABSTRACT Click reactions are a highly versatile class of reactions that produce a diverse range of products. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) click reactions require an azide and a terminal alkyne and produce a coupled product that is “clicked” through a triazole ring that can have a variety of substituents. In this work, bromo-terminated phosphonate films on copper oxide surfaces were explored as the platform for click coupling, as the terminal azide needed for the reaction can be generated through an in situ SN2 reaction with a terminal bromo group. The reactions were characterized using model reactions in solution before being conducted on modified copper oxide surfaces. -

Synthesis of Protected 2′-Deoxy-2′-Fluoro-Β-D-Arabinonucleosides
Synthesis of Protected UNIT 1.7 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides This unit describes in detail the preparation of protected 2′-deoxy-2′-fluoroarabinonu- cleosides. These building blocks are required for the synthesis of 2′-deoxy-2′-fluoroarabi- nonucleic acid (2′F-ANA), an oligonucleotide analog exhibiting very promising antisense properties (Damha et al., 1998; Wilds and Damha, 2000; Lok et al., 2002). The preparation of phosphoramidites from these building blocks and the synthesis of 2′F-ANA are described in UNIT 4.15. SYNTHESIS AND CHARACTERIZATION OF N2-ISOBUTYRYL-9- BASIC [2-DEOXY-2-FLUORO-5-O-(4-METHOXYTRITYL)-β-D- PROTOCOL 1 ARABINOFURANOSYL]GUANINE Synthesis of araF-G (S.6; Figure 1.7.3) was accomplished via the condensation of 2,6-dichloropurine with either 2-deoxy-2-fluoro-1,3,5-tri-O-benzoyl-α-D-arabinofura- nose (S.2; Figure 1.7.1) or 2-deoxy-2-fluoro-3,5-di-O-benzoyl-α-D-arabinofuranosyl bromide (S.3) as a key chemical step (Figure 1.7.2). Starting from S.3 gives a higher yield of S.4 and simplifies its purification. The versatile intermediate N9-β-glycoside (S.4) was transformed into araF-G in three steps (Figure 1.7.3). Treatment of S.4 with NaN3 in ethanol gave the 2,6-diazido derivative (Wower et al., 1994). This product was then subjected to reduction with SnCl2 in a mixture of dichloromethane/methanol to give its 2,6-diamino derivative (Tennila et al., 2000). Standard debenzoylation and deamination gave araF-G. -

Carbon–Fluorine Bond Formation
Carbon–Fluorine Bond Formation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Furuya, Takeru, Christian A. Kuttruff, and Tobias Ritter. 2008. Carbon–fluorine bond formation. Current Opinion in Drug Discovery and Development 11(6): 803–819. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:8301597 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Open Access Policy Articles, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#OAP Carbon–Fluorine Bond Formation Takeru Furuya, Christian A. Kuttruff, and Tobias Ritter* Department of Chemistry and Chemical Biology Harvard University 12 Oxford Street Cambridge, MA 02138 [email protected] Phone 617 496 0750 Fax 617 496 4591 1 Abstract We present a selection of carbon–fluorine bond formations that have been developed in the recent past. An overview of the most common fluorination reagents is followed by fluorination reactions organized by reactivity. We have distinguished between nucleophilic and electrophilic fluorinations as well as aliphatic and aromatic fluorinations. Each section is divided into more specific reaction classes and examples for syntheses of pharmaceuticals, 18F-radiolabeling, and mechanistic investigations are provided. Keywords Fluorination; carbon–fluorine bond formation; nucleophilic fluorination; electrophilic fluorination; fluorinating -
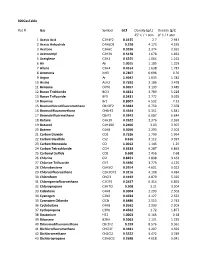
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Oxidation Trifluoride (DAST) Selectfluor™
Vol. 5 No. 1 Synthetic Methods Iodotoluene difluoride Diethylaminosulfur Oxidation trifluoride (DAST) Selectfluor™ Ishikawa’s Reagent Other Reagents for Fluorination Dess–Martin Periodinane Bis(pyridine)iodonium tetrafluoroborate Magnesium bis(monoperoxyphthalate) hexahydrate Chloramine-T Other Reagents for Oxidation Advancing Science 2 Introduction Oxidation is one of the most common transformations in all of organic chemistry. As a result, the number of oxidizing agents and reactions at the research chemist’s disposal number in the hundreds; Sigma-Aldrich is proud to offer a new series of ChemFiles—called however, few provide extended utility across a broad range of Synthetic Methods—to our Organic Chemistry and Drug Discovery substrates while remaining able to selectively oxidize one group customers. Each piece will highlight a specific motif or selected over another. Fewer yet remain effective under mild conditions. At organic transformation, and the range of products Sigma-Aldrich Sigma-Aldrich, we are committed to being your preferred supplier offers in this area. Our first installment focuses on oxidation for reagents used in oxidation—and we’d like to highlight some chemistry. In the future, we will highlight other synthetic methods new, popular, and versatile oxidation reagents in this ChemFile. such as asymmetric synthesis and reduction. We trust you will find these pieces useful, and we welcome your comments for Listed on the following pages is a selection of oxidation reagents future installments. If you cannot find a product for your specific available from Sigma-Aldrich. For a complete listing of oxidation research in organic synthesis or drug discovery, we encourage your reagents, please visit sigma-aldrich.com/oxidation. -

H-Phosphonates: Versatile Synthetic Precursors to Biologically Active Phosphorus Compounds*
Pure Appl. Chem., Vol. 79, No. 12, pp. 2217–2227, 2007. doi:10.1351/pac200779122217 © 2007 IUPAC H-Phosphonates: Versatile synthetic precursors to biologically active phosphorus compounds* Adam Kraszewski1,‡ and Jacek Stawinski1,2,‡ 1Institute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-704 Poznan, Poland; 2Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, S-10691 Stockholm, Sweden Abstract: In this review, a short account of H-phosphonate chemistry and its application to the synthesis of biologically important phosphates and their analogs is given. Keywords: H-phosphonates; phosphate analogs; biological activity; nucleic acids; drugs. INTRODUCTION Located at the crossroad of various bioinformation exchange pathways, phosphorus-containing com- pounds play a key role in living organisms as carriers of genetic information and important signalling, regulatory, energy transfer, and structural compounds [1]. Due to this pivotal role, biologically impor- tant phosphorus compounds have become therapeutic targets in various modern medicinal techniques, such as antisense [2] and antigene [3] approaches to modulation gene expression, or a gene silencing technique using short interfering RNA (siRNA) [4]. To maximize the desired biological effect and to minimize cytotoxicity, a biologically active com- pound has to have very precisely adjusted chemical and pharmacokinetic properties. This can be achieved by, e.g., changing pKa values of the ionizeable functions, changing electronegativity of the substituents, their size, hydrophobicity, etc. These features can also influence transport of a drug across cell membranes, its preferential degradation in normal or activation in the neoplastic or virus-infected cells, and can contribute to the overall efficiency of the drug. A prominent example here are antiviral pronucleotides in which the phosphate moiety is masked with alkyl or aryl groups that are removed chemically and enzymatically in cells to produce an active antiviral nucleotide [5]. -

The Hydrolysis of Phosphinates and Phosphonates: a Review
molecules Review The Hydrolysis of Phosphinates and Phosphonates: A Review Nikoletta Harsági and György Keglevich * Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary; [email protected] * Correspondence: [email protected]; Tel.: +36-1-463-1111 (ext. 5883) Abstract: Phosphinic and phosphonic acids are useful intermediates and biologically active com- pounds which may be prepared from their esters, phosphinates and phosphonates, respectively, by hydrolysis or dealkylation. The hydrolysis may take place both under acidic and basic conditions, but the C-O bond may also be cleaved by trimethylsilyl halides. The hydrolysis of P-esters is a challenging task because, in most cases, the optimized reaction conditions have not yet been explored. Despite the importance of the hydrolysis of P-esters, this field has not yet been fully surveyed. In order to fill this gap, examples of acidic and alkaline hydrolysis, as well as the dealkylation of phosphinates and phosphonates, are summarized in this review. Keywords: hydrolysis; dealkylation; phosphinates; phosphonates; P-acids 1. Introduction Phosphinic and phosphonic acids are of great importance due to their biological activity (Figure1)[ 1]. Most of them are known as antibacterial agents [2,3]. Multidrug- resistant (MDR) and extensively drug-resistant (XDR) pathogens may cause major problems Citation: Harsági, N.; Keglevich, G. in the treatment of bacterial infections. However, Fosfomycin has remained active against The Hydrolysis of Phosphinates and both Gram-positive and Gram-negative MDR and XDR bacteria [2]. Acyclic nucleoside Phosphonates: A Review. Molecules phosphonic derivatives like Cidofovir, Adefovir and Tenofovir play an important role 2021, 26, 2840. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Discovery of Phosphonic Acid Natural Products by Mining the Genomes of 10,000 Actinomycetes
Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes Kou-San Jua, Jiangtao Gaoa, James R. Doroghazia, Kwo-Kwang A. Wanga, Christopher J. Thibodeauxa, Steven Lia, Emily Metzgera, John Fudalaa, Joleen Sua, Jun Kai Zhanga,b, Jaeheon Leea, Joel P. Cionia,b, Bradley S. Evansa, Ryuichi Hirotaa,c, David P. Labedad, Wilfred A. van der Donka,e,1, and William W. Metcalfa,b,1 aCarl R. Woese Institute for Genomic Biology, University of Illinois, Urbana, IL 61801; bDepartment of Microbiology, University of Illinois, Urbana, IL 61801; cDepartment of Molecular Biotechnology, Graduate School of Advanced Sciences of Matter, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima, Hiroshima 739-8530, Japan; dBacterial Foodborne Pathogens and Mycology Research, US Department of Agriculture, Agricultural Research Service, National Center for Agricultural Utilization Research, Peoria, IL 61604; and eDepartment of Chemistry and Howard Hughes Medical Institute, University of Illinois, Urbana, IL 61081 Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved July 31, 2015 (received for review January 14, 2015) Although natural products have been a particularly rich source of generally adopted. If we hope to revitalize the use of natural human medicines, activity-based screening results in a very high products in the pharmaceutical industry, genome mining must be rate of rediscovery of known molecules. Based on the large number shown to be a high-throughput discovery process complementary of natural product biosynthetic genes in microbial genomes, many or superior to existing methods (13, 14). Here, we demonstrate the have proposed “genome mining” as an alternative approach for feasibility of this approach in a campaign to identify the full ge- discovery efforts; however, this idea has yet to be performed ex- netic repertoire of phosphonic acid natural products encoded by a perimentally on a large scale. -

Addition of A-Lithioalkane Phosphonates to Nitriles, Synthesis of ;3-Keto Phosphonate and Enamine Phosphonates
Communications to the Editor Bull. Korean Chem. Soc., Vol. 10, No. 6, 1989 613 Addition of a-Lithioalkane Phosphonates to Nitriles, Synthesis of ;3-Keto Phosphonate and Enamine Phosphonates Kilsung Lee and Dong Young Oh * Department of Chemistry, Korea Advanced Institute of Science and Technology, Seoul 130-650 Received August 28, 1989 ^-Keto phosphonates bearing a a -hydrogen are in 0 0 N" 0 NH teresting for the conversion under Wittig-Horner condition (EtO)J다% .1,叶如닌T (EtO)5PCH--CPh 二二 (EtO)?PCH-CPh into the corresponding ff,/3-unsaturated ketones.1 fi-Keto £ S 2. PhCN £ c phosphonates fully substituted at the a-carbon do not find 4 5 such frequent use as synthetic intermediates, but because the geometry and spatial demands of phosphorus group are comparable to those of quaternary carbon, the isosteric re placement of carbon with phosphorus has been studied in biologically active molecules.2 In marked contrast to a number of investigations that in corporate phosphonate reagents or focus on the biological ac tivities of phosphonates, r이atively little work has appeared describing general new synthesis of this functionality (par ticularly in case of fully substituted /9-keto phosphonate).3 Recently developed reaction of dialkyl phosphoro chloridites with a -hydroxy carbonyl compounds in the presence of a a-hydrogen dimerized products were obtained as side pro Lewis acid is a good candidate for fully substituted g-ket。 ducts, as previously reported by G. Sumrell.5*2 In the case of phosphonates.4 However, its process is limited due primary an phenyl or thiophenyl substituent on the carbon to the to the availability of starting material. -

Cyclophostin, (±)-Cyclipostin P, Their Phosphonate Analogs and New
University of Missouri, St. Louis IRL @ UMSL Dissertations UMSL Graduate Works 8-2-2011 Synthesis of (±)-Cyclophostin, (±)-Cyclipostin P, Their hoP sphonate Analogs and New Chemistry of Vinyl Phosphonates Raj Kumar Malla University of Missouri-St. Louis, [email protected] Follow this and additional works at: https://irl.umsl.edu/dissertation Part of the Chemistry Commons Recommended Citation Malla, Raj Kumar, "Synthesis of (±)-Cyclophostin, (±)-Cyclipostin P, Their hospP honate Analogs and New Chemistry of Vinyl Phosphonates" (2011). Dissertations. 416. https://irl.umsl.edu/dissertation/416 This Dissertation is brought to you for free and open access by the UMSL Graduate Works at IRL @ UMSL. It has been accepted for inclusion in Dissertations by an authorized administrator of IRL @ UMSL. For more information, please contact [email protected]. Synthesis of (±)-Cyclophostin, (±)-Cyclipostin P, Their Phosphonate Analogs and New Chemistry of Vinyl Phosphonates by Raj Kumar Malla A Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Chemistry) University of Missouri-St. Louis August, 2011 Committee Prof. Christopher D. Spilling (Chair and Advisor) Prof. Alexei V. Demchenko Prof. Cynthia M. Dupureur Prof. Eike B. Bauer Abstract Synthesis of (±)-Cyclophostin, (±)-Cyclipostin P, Their Phosphonate Analogs and New Chemistry of Vinyl Phosphonates Raj Kumar Malla Doctor of Philosophy University of Missouri-St. Louis Prof. Christopher D. Spilling, Advisor This work is divided into 2 parts: a) total synthesis of (±)-cyclophostin, (±)-cyclipostin P and their monocyclic analogs b) development of a metathesis based methodology for transformation of vinyl phosphonates. Cyclophostin (1) and cyclipostins (2) are bioactive natural bicyclic organophosphates. -

Wittig and Wittig-Horner Reactions Under Phase Transfer Catalysis
CEJC 4(2003)491{542 Review article Wittigand Wittig-Horner reactions underphase transfer catalysis conditions AureliaPascariu, Gheorghe Ilia ¤,AlinaBora, Smaranda Iliescu, AdrianaPopa, Gheorghe Dehelean and Liliana Pacureanu Institute ofChemistry, Romanian A cademy, 24M. Viteazul Bv., R300223Timisoara, Romania Received 2April 2003;accepted 11August 2003 Abstract: Wittig andWittig-Horner reactions arefavorite tools in preparative organic chemistry.Theseole nation methodsenjoy widespread andrecognition becauseof their simplicity,convenience, ande¯ ciency.Phase transfer catalysis (PTC) is avery important methodin synthetic organicchemistry havingmany advantages over conventional, homogenous reaction procedures. Inthis paper, weattempt to summarizethe aspects concerningWittig andWittig-Horner reactions that take place under phasetransfer catalysis conditions. c Central EuropeanScience Journals. All rights reserved. ® Keywords: Wittig reactions, Wittig-Horner reactions, phasetr ansfercatalysis 1Introduction Thanks to numerous theoretical and practical implications,the modern chemistry of phosphorus draws increasing attention. Many scientists havededicated their work inves- tigating someof the unexplained aspects of organophosphorus compounds. They have synthesized new compounds and haveused them inthe new ¯eldof ¯ne organic synthesis or in order to clarifysome of the reaction mechanisms. Although vast literature concerning phase transfer catalysisis available, information concerning the Wittig and Wittig-Horner reactions are treated separately,not