CLINICAL STUDY PROTOCOL X16069 Phase I/II Study of Bendamustine and IXAZOMIB (MLN9708) Plus Dexamethasone in Relapsed/Refractory Multiple Myeloma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PPE Requirements Hazardous Drug Handling
This document’s purpose is only to provide general guidance. It is not a definitive interpretation for how to comply with DOSH requirements. Consult the actual NIOSH hazardous drugs list and program regulations in entirety to understand all specific compliance requirements. Minimum PPE Required Minimum PPE Required Universal (Green) - handling and disposed of using normal precautions. PPE Requirements High (Red) - double gloves, gown, eye and face protection in Low (Yellow) - handle at all times with gloves and appropriate engineering Hazardous Drug Handling addition to any necessary controls. engineering controls. Moderate (Orange) -handle at all times with gloves, gown, eye and face protection (with splash potential) and appropirate engineering controls. Tablet Open Capsule Handling only - Contained Crush/Split No alteration Crush/Split Dispensed/Common Drug Name Other Drug Name Additional Information (Formulation) and (NIOSH CATEGORY #) Minimum PPE Minimum PPE Minimum PPE Minimum PPE Required required required required abacavir (susp) (2) ziagen/epzicom/trizivir Low abacavir (tablet) (2) ziagen/epzicom/trizivir Universal Low Moderate acitretin (capsule) (3) soriatane Universal Moderate anastrazole (tablet) (1) arimidex Low Moderate High android (capsule) (3) methyltestosterone Universal Moderate apomorphine (inj sq) (2) apomorphine Moderate arthotec/cytotec (tablet) (3) diclofenac/misoprostol Universal Low Moderate astagraf XL (capsule) (2) tacrolimus Universal do not open avordart (capsule) (3) dutasteride Universal Moderate azathioprine -

Handling of Hazardous Drugs Risk Prevention by Personal Protective Equipment Handling of Hazardous Drugs
HANDLING OF HAZARDOUS DRUGS RISK PREVENTION BY PERSONAL PROTECTIVE EQUIPMENT HANDLING OF HAZARDOUS DRUGS INTRODUCTION CONTENT The expression “antineoplastic drug” (ANPD) is often used synonymously together with “cytostatics” or Introduction 2 “chemotherapeutics”, however, these terms normally Definition of Risks 4 describe an overarching category to which other drug- classes belong. These types of drugs belong to drug- Causes for Risks 10 specialties summarized under the term “Hazardous Consequences 14 Drugs”, according to the CDC’s (Centers for Disease Control and Prevention) NIOSH4 alert in 2004. The Preventive Strategies 16 term ANPD describes in general the activity of these Risk Prevention 26 drugs against a neoplasm, characterizing an abnormal growth of tissue. In a recent systematic review and Literature 28 meta-analysis of the literature5 the expression “ANPD” Mandatory Information 31 is used in a general manner, therefore it is applied in this review, too. INTRODUCTION Antineoplastic drugs (ANPD) have been introduced ANPDs represent a broad and non-homogenous group for cancer treatment since the 1940s. More than of chemicals with a variety of structures, origins, ac- 12 million patients are treated with ANPDs each tivities and effects at the cellular level. They are cate- year. Nowadays the number of cancer diagnoses gorized according to their specific potential of toxicity is continuously increasing. or to their mechanisms of action, described in more detail in the chapter “Definition of risks”. Currently, the This brochure addresses the hazardous effects of anti- list encompassing ANPDs used in daily clinical practice neoplastic drugs, the importance of risk assessment contains more than 115 special drugs1. and standard precautions of personal protection as recommended by the 2004-NIOSH (National Institute During the 1970’s first concerns with respect to toxic for Occupational Safety and Health)-Alert and corre- side effects were raised further to cases that had been 1, 2, 3 sponding updates in 2010 / 2012 and 2016. -

Characterization and Separation of Platinum-Based Antineoplastic
separations Article Characterization and Separation of Platinum-Based Antineoplastic Drugs by Zwitterionic Hydrophilic Interaction Liquid Chromatography (HILIC)–Tandem Mass Spectrometry, and Its Application in Surface Wipe Sampling Stefano Dugheri 1,* , Nicola Mucci 2 , Enrico Mini 3, Donato Squillaci 2 , Giorgio Marrubini 4 , Gianluca Bartolucci 5 , Elisabetta Bucaletti 2, Giovanni Cappelli 2, Lucia Trevisani 2 and Giulio Arcangeli 2 1 Industrial Hygiene and Toxicology Laboratory, Occupational Medicine Unit, Careggi University Hospital, 50134 Florence, Italy 2 Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy; nicola.mucci@unifi.it (N.M.); donato.squillaci@unifi.it (D.S.); elisabetta.bucaletti@unifi.it (E.B.); giovanni.cappelli@unifi.it (G.C.); lucia.trevisani@unifi.it (L.T.); giulio.arcangeli@unifi.it (G.A.) 3 Department of Health Sciences, University of Florence, 50134 Florence, Italy; enrico.mini@unifi.it 4 Department of Drug Sciences, University of Pavia, Via Taramelli 12, 27100 Pavia, Italy; [email protected] 5 Department of Neurosciences, Psychology, Drug Research and Child Health, University of Florence, 50019 Sesto Fiorentino, Italy; gianluca.bartolucci@unifi.it Citation: Dugheri, S.; Mucci, N.; * Correspondence: stefano.dugheri@unifi.it Mini, E.; Squillaci, D.; Marrubini, G.; Bartolucci, G.; Bucaletti, E.; Cappelli, Abstract: Platinum-based antineoplastic drugs (PtADs) are among the most important and used G.; Trevisani, L.; Arcangeli, G. families of chemotherapy drugs, which, -

ASHP Guidelines on Handling Hazardous Drugs
132 Drug Distribution and Control: Preparation and Handling–Guidelines ASHP Guidelines on Handling Hazardous Drugs ASHP published its first guidance on hazardous drugs (HDs) Because newer studies have shown that contamination in 1983 as part of the 1983–84 ASHP Practice Spotlight: Safe is widespread in healthcare settings and that more workers Handling of Cytotoxic Drugs.1,2 This was followed by tech- than previously thought are exposed, these guidelines should nical assistance bulletins in 1985 and 1990 and the ASHP be implemented wherever HDs are received, stored, pre- Guidelines on Handling Hazardous Drugs in 2006.3-5 The pared, transported, administered, or disposed.8-11 2006 guidelines were created to harmonize with the National Comprehensive reviews of the literature covering an- Institute for Occupational Safety and Health (NIOSH) Alert: ecdotal and case reports of surface contamination, worker ex- 6,9,12 Preventing Occupational Exposure to Antineoplastic and posure, and risk assessment are available from NIOSH, Other Hazardous Drugs in Health Care Settings issued in the Occupational Safety and Health Administration 13,14 15-20 2004.6 The ASHP 2006 HD guidelines were current to 2005. (OSHA), and individual authors. The primary goal In 2007, the United States Pharmacopeial Convention of this document is to provide recommendations for the safe revised United States Pharmacopeia (USP) chapter 797 handling of HDs. These guidelines represent the research (Pharmaceutical Compounding—Sterile Preparations)7 to and recommendations of many groups and individuals who harmonize with the NIOSH 2004 Alert. It became effective have worked tirelessly over decades to reduce the potential May 1, 2008, establishing many of the NIOSH recommenda harmful effects of HDs on healthcare workers. -

Targeted Therapies and Immunotherapy: General Principles (Oncology) – CE
Targeted Therapies and Immunotherapy: General Principles (Oncology) – CE ALERT Don appropriate personal protective equipment (PPE) based on the patient’s signs and symptoms and indications for isolation precautions. Refer to Oncology Nursing Society (ONS) interim guidelines for PPE recommendations during an emergent shortage of PPE (e.g., pandemic).13 Hypersensitivity reactions, such as anaphylaxis, may occur with targeted therapy and immunotherapy; therefore, frequent assessments and monitoring are required. Only qualified physicians, physician assistants, advanced practice registered nurses (APRNs), or nurses with demonstrated competency administer antineoplastic therapies. Refer to the professional’s regulatory scope of practice and the organization’s practice. Take steps to eliminate interruptions and distractions during medication preparation. OVERVIEW Normal cell reproduction, growth, and apoptosis are controlled by complex signaling pathways at the extracellular and intracellular levels. Malfunctioning of these pathways occurs in malignancies, leading to increased proliferation, tissue invasion, metastases, and apoptosis inhibition.18 Development of targeted therapies overcame the lack of selectivity associated with conventional antineoplastic therapy by targeting specific protein pathways.5 Although these pathways can be present in normal tissue, they are overexpressed or mutated in cancerous tissue.1 Cancer immunotherapy represents precision medicine, which helps the immune system fight cancer and offers a type of targeted or personalized therapy.4,11 In comparison, traditional cytotoxic chemotherapy attacks both malignant and nonmalignant cells, causing disruption of the cell cycle and other cell functions. Targeted therapy may not be more or less effective than traditional antineoplastic therapies, but it offers a unique approach to the treatment of cancer and other diseases and has toxicities different from those of traditional antineoplastic therapy. -

Dactinomycin for INJECTION, USP
Dactinomycin FOR INJECTION, USP 10 DIGIT NDC STRENGTH SIZE WHOLESALE NUMBERS WEB LISTING 39822-2100-2 ABC 10229241 500 mcg 1 Cardinal 5557707 11 DIGIT NDC (0.5 mg) per vial vial McKesson 3975687 39822-2100-02 Morris Dickson 772301 OTHER INFORMATION PRESERVATIVE FREE LATEX FREE STOPPER CONTROLLED ROOM TEMPERATURE STORAGE - - - BARCODED FOR PATIENT SAFETY Dactinomycin FOR INJECTION, USP HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use • Regional Perfusion in Locally Recurrent and Locoregional Solid Malignancies: Dactinomycin for Injection safely and effectively. See full prescribing • Lower Extremity or Pelvis: The recommend dose is 50 mcg/kg once with information for Dactinomycin for Injection. melphalan. (2.6) ◦ Upper Extremity: The recommended dose is 35 mcg/kg once with melphalan. (2.6) Dactinomycin for Injection for intravenous use Initial U.S. Approval: 1964 DOSAGE FORMS AND STRENGTHS For injection: 500 mcg as a lyophilized powder in a single-dose vial. (3) RECENT MAJOR CHANGES • Dosage and Administration, Recommended Dosage for Wilms Tumor (2.1) CONTRAINDICATIONS 8/2018 None. (4) • Dosage and Administration, Recommended Dosage for Ewing Sarcoma (2.3) 8/2018 WARNINGS AND PRECAUTIONS • Secondary Malignancy or Leukemia: Increased risk of secondary malignancies INDICATIONS AND USAGE following treatment. (5.1) Dactinomycin for injection is an actinomycin indicated for the treatment of: • Veno-occlusive Disease: Can cause severe or fatal VOD. Monitor for elevations in • adult and pediatric patients with Wilms tumor, as part of a multi-phase, AST, ALT, total bilirubin, hepatomegaly, weight gain, or ascites. Consider delaying combination chemotherapy regimen. (1.1) next dose. -

Providing Care for the Person Receiving Cancer Biological And
MODULE SIX – PART FOUR Providing care for the person receiving cancer biological and molecular targeted therapies Overview Estimated time to complete The aim of this module is to develop the ability of 20 hours the beginning specialist cancer nurse to demonstrate competence across all domains of practice when caring for the person receiving biological or molecular targeted Objectives therapies for cancer. On completion of this supporting resource, you should be able to: Key concepts 1. Perform a comprehensive health assessment on a person prior to, during and following biological and The key concepts associated with providing care for the molecular targeted therapies. person receiving cancer biological and molecular targeted 2. Analyse clinical, psychological and social data to therapies include: formulate and implement an individualised plan of care • Factors influencing selection of biological and molecular for the person having biological and molecular targeted targeted therapies for treatment of cancer. therapies. • Experience and impact of biological and molecular 3. Demonstrate delivery of effective nursing care to targeted therapies on various health domains. prevent, detect and manage early and late effects • Prevention, detection, and management of common associated with biological and molecular targeted health alterations experienced by people receiving therapies. biological and molecular targeted therapies. 4. Demonstrate effective educational strategies in • Management of an adverse reaction to cancer biological providing individualised information to the person and molecular targeted therapies – demonstration of having biological and molecular targeted therapies. competence within ICAT. 5. Demonstrate safe practice in the care of the person receiving cytotoxic agents. Learning activities 6. Demonstrate competence in the administration of biological and molecular targeted therapies. -

Prescribing Information | VELCADE® (Bortezomib)
HIGHLIGHTS OF PRESCRIBING INFORMATION Hypotension: Use caution when treating patients taking These highlights do not include all the information needed to antihypertensives, with a history of syncope, or with dehydration. use VELCADE safely and effectively. See full prescribing (5.2) information for VELCADE. Cardiac Toxicity: Worsening of and development of cardiac VELCADE® (bortezomib) for injection, for subcutaneous or failure has occurred. Closely monitor patients with existing heart intravenous use disease or risk factors for heart disease. (5.3) Initial U.S. Approval: 2003 Pulmonary Toxicity: Acute respiratory syndromes have ------------------------RECENT MAJOR CHANGES-------------------------- occurred. Monitor closely for new or worsening symptoms and consider interrupting VELCADE therapy. (5.4) Warnings and Precautions, Posterior Reversible Encephalopathy Syndrome: Consider MRI Thrombotic Microangiopathy (5.10) 04/2019 imaging for onset of visual or neurological symptoms; ------------------------INDICATIONS AND USAGE-------------------------- discontinue VELCADE if suspected. (5.5) VELCADE is a proteasome inhibitor indicated for: Gastrointestinal Toxicity: Nausea, diarrhea, constipation, and treatment of adult patients with multiple myeloma (1.1) vomiting may require use of antiemetic and antidiarrheal medications or fluid replacement. (5.6) treatment of adult patients with mantle cell lymphoma (1.2) Thrombocytopenia and Neutropenia: Monitor complete blood ----------------------DOSAGE AND ADMINISTRATION--------------------- -

Velcade Label
HIGHLIGHTS OF PRESCRIBING INFORMATION • Hypotension can occur. Caution should be used when treating These highlights do not include all the information needed to use patients receiving antihypertensives, those with a history of syncope, VELCADE safely and effectively. See full prescribing information and those who are dehydrated. (5.3) for VELCADE. • Patients with risk factors for, or existing heart disease, should be closely monitored. (5.4) VELCADE (bortezomib) for Injection • Acute diffuse infiltrative pulmonary disease has been reported. (5.5) Initial US Approval: 2003 • Nausea, diarrhea, constipation, and vomiting have occurred and may -------------------------RECENT MAJOR CHANGES---------------------- require use of antiemetic and antidiarrheal medications or fluid Patients with Renal Impairment (8.6) 10/2007 replacement. (5.7) Indications and Usage, Multiple Myeloma (1.1) 06/2008 • Thrombocytopenia or neutropenia can occur; complete blood counts ------------------------INDICATIONS AND USAGE------------------------- should be regularly monitored throughout treatment. (5.8) VELCADE is a proteasome inhibitor indicated for: • Tumor Lysis Syndrome (5.9), Reversible Posterior • treatment of patients with multiple myeloma (1.1) Leukoencephalopathy Syndrome (5.6), and acute hepatic failure • treatment of patients with mantle cell lymphoma who have received at (5.10) have been reported. least 1 prior therapy (1.2) --------------------------ADVERSE REACTIONS------------------------- -------------------DOSAGE AND ADMINISTRATION-------------------- 2 Most commonly reported adverse reactions (incidence ≥30%) in The recommended dose of VELCADE is 1.3 mg/m administered as a 3 clinical studies include asthenic conditions, diarrhea, nausea, to 5 second bolus intravenous injection. (2.1, 2.3) constipation, peripheral neuropathy, vomiting, pyrexia, Dose adjustment may be used to manage adverse events that occur thrombocytopenia, psychiatric disorders, anorexia and decreased during treatment (2.2, 2.4) appetite, neutropenia, neuralgia, leukopenia and anemia. -

USP 800-Hazardous Drug Handling in Healthcare Settings
2017 Authorized reprint for individual use only. Must be downloaded with registration directly from www.usp.org USP General Chapter <800> Hazardous Drugs – Handling in Healthcare Settings Reprinted from USP 40—NF 35, Second Supplement (2017) Links For Supplemental Resources Information on USP General Chapter <800> USP General Chapter <800> FAQs USP General Chapter <800> Education Courses Sign up for USP updates This text is a courtesy copy of General Chapter <800> Hazardous Drugs – Handling in Healthcare Settings, intended to be used as an informational tool and resource only. Please refer to the current edition of the USP-NF for official text. This chapter alone is not sufficient for a comprehensive approach to safe handling of hazardous drugs. Additional chapters are required for complete implementation; see USP Compounding Compendium or USP-NF. Accessed from 10.10.244.113 by uspstaff on Tue Sep 19 09:21:47 EDT 2017 Errata to First Supplement to USP 40–NF 35 Physical Tests / á800ñ Hazardous Drugs 1 Add the following: á800ñ HAZARDOUS DRUGS—HANDLING IN HEALTHCARE SETTINGS (Chapter to become official July 1, 2018.) Refer to USP Notice of Intent to Revise (9/29/2017) 1. INTRODUCTION AND SCOPE This chapter describes practice and quality standards for handling hazardous drugs (HDs) to promote patient safety, worker safety, and environmental protection. Handling HDs includes, but is not limited to, the receipt, storage, compounding, dis- pensing, administration, and disposal of sterile and nonsterile products and preparations. This chapter applies to all healthcare personnel who handle HD preparations and all entities that store, prepare, transport, or administer HDs (e.g., pharmacies, hospitals and other healthcare institutions, patient treatment clinics, physicians' practice fa- cilities, or veterinarians' offices). -
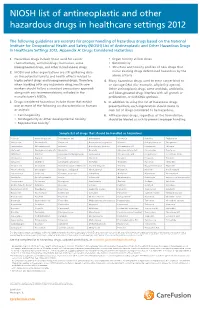
NIOSH List of Antineoplastic and Other Hazardous Drugs in Healthcare Settings 2012
NIOSH list of antineoplastic and other hazardous drugs in healthcare settings 2012 The following guidelines are excerpts for proper handling of hazardous drugs based on the National Institute for Occupational Health and Safety (NIOSH) List of Antineoplastic and Other Hazardous Drugs in Healthcare Settings 2012, Appendix A: Drugs Considered Hazardous 1. Hazardous drugs include those used for cancer • Organ toxicity at low doses* chemotherapy, antiviral drugs, hormones, some • Genotoxicity** bioengineered drugs, and other miscellaneous drugs. • Structure and toxicity profiles of new drugs that 2. NIOSH and other organizations are still gathering data mimic existing drugs determined hazardous by the on the potential toxicity and health effects related to above criteria highly potent drugs and bioengineered drugs. Therefore, 4. Many hazardous drugs used to treat cancer bind to when working with any hazardous drug, health care or damage DNA (for example, alkylating agents). workers should follow a standard precautions approach Other antineoplastic drugs, some antivirals, antibiotics, along with any recommendations included in the and bioengineered drugs interfere with cell growth or manufacturer’s MSDSs. proliferation, or with DNA synthesis. 3. Drugs considered hazardous include those that exhibit 5. In addition to using the list of hazardous drugs one or more of the following six characteristics in humans presented here, each organization should create its or animals: own list of drugs considered to be hazardous. • Carcinogenicity 6. All hazardous -

CYTOTOXIC and NON-CYTOTOXIC HAZARDOUS MEDICATIONS
CYTOTOXIC and NON-CYTOTOXIC HAZARDOUS MEDICATIONS1 CYTOTOXIC HAZARDOUS MEDICATIONS NON-CYTOTOXIC HAZARDOUS MEDICATIONS Altretamine IDArubicin Acitretin Iloprost Amsacrine Ifosfamide Aldesleukin Imatinib 3 Arsenic Irinotecan Alitretinoin Interferons Asparaginase Lenalidomide Anastrazole 3 ISOtretinoin azaCITIDine Lomustine Ambrisentan Leflunomide 3 azaTHIOprine 3 Mechlorethamine Bacillus Calmette Guerin 2 Letrozole 3 Bleomycin Melphalan (bladder instillation only) Leuprolide Bortezomib Mercaptopurine Bexarotene Megestrol 3 Busulfan 3 Methotrexate Bicalutamide 3 Methacholine Capecitabine 3 MitoMYcin Bosentan MethylTESTOSTERone CARBOplatin MitoXANtrone Buserelin Mifepristone Carmustine Nelarabine Cetrorelix Misoprostol Chlorambucil Oxaliplatin Choriogonadotropin alfa Mitotane CISplatin PACLitaxel Cidofovir Mycophenolate mofetil Cladribine Pegasparaginase ClomiPHENE Nafarelin Clofarabine PEMEtrexed Colchicine 3 Nilutamide 3 Cyclophosphamide Pentostatin cycloSPORINE Oxandrolone 3 Cytarabine Procarbazine3 Cyproterone Pentamidine (Aerosol only) Dacarbazine Raltitrexed Dienestrol Podofilox DACTINomycin SORAfenib Dinoprostone 3 Podophyllum resin DAUNOrubicin Streptozocin Dutasteride Raloxifene 3 Dexrazoxane SUNItinib Erlotinib 3 Ribavirin DOCEtaxel Temozolomide Everolimus Sirolimus DOXOrubicin Temsirolimus Exemestane 3 Tacrolimus Epirubicin Teniposide Finasteride 3 Tamoxifen 3 Estramustine Thalidomide Fluoxymesterone 3 Testosterone Etoposide Thioguanine Flutamide 3 Tretinoin Floxuridine Thiotepa Foscarnet Trifluridine Flucytosine Topotecan Fulvestrant