Quantum Dot Metrics for Preparative Chemistry and Fluorescence Applications Yi Shen University of South Carolina
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alfa Laval Black and Grey List, Rev 14.Pdf 2021-02-17 1678 Kb
Alfa Laval Group Black and Grey List M-0710-075E (Revision 14) Black and Grey list – Chemical substances which are subject to restrictions First edition date. 2007-10-29 Revision date 2021-02-10 1. Introduction The Alfa Laval Black and Grey List is divided into three different categories: Banned, Restricted and Substances of Concern. It provides information about restrictions on the use of Chemical substances in Alfa Laval Group’s production processes, materials and parts of our products as well as packaging. Unless stated otherwise, the restrictions on a substance in this list affect the use of the substance in pure form, mixtures and purchased articles. - Banned substances are substances which are prohibited1. - Restricted substances are prohibited in certain applications relevant to the Alfa Laval group. A restricted substance may be used if the application is unmistakably outside the scope of the legislation in question. - Substances of Concern are substances of which the use shall be monitored. This includes substances currently being evaluated for regulations applicable to the Banned or Restricted categories, or substances with legal demands for monitoring. Product owners shall be aware of the risks associated with the continued use of a Substance of Concern. 2. Legislation in the Black and Grey List Alfa Laval Group’s Black and Grey list is based on EU legislations and global agreements. The black and grey list does not correspond to national laws. For more information about chemical regulation please visit: • REACH Candidate list, Substances of Very High Concern (SVHC) • REACH Authorisation list, SVHCs subject to authorization • Protocol on persistent organic pollutants (POPs) o Aarhus protocol o Stockholm convention • Euratom • IMO adopted 2015 GUIDELINES FOR THE DEVELOPMENT OF THE INVENTORY OF HAZARDOUS MATERIALS” (MEPC 269 (68)) • The Hong Kong Convention • Conflict minerals: Dodd-Frank Act 1 Prohibited to use, or put on the market, regardless of application. -

The East African Sub-Regional Project
UNEPIUNDP/DUTCH Joint Project on Environmental Law UNEP and Institutions in Africa THE EAST AFRICAN SUB-REGIONAL PROJECT DEVELOPMENT AND HARMONISATION OF ENVIRONMENTAL LAWS VOLUME 4 DECEMBER 1999 ,44 UNEP/UNDP/DUTCH Joint Project on Environmental Law and Institutions in Africa UNEP UWD TILE EAST AFRICAN SUB-REGIONAL PROJECT DEVELOPMENT AND HARMONISATION OF ENVIRONMENTAL LAWS VOLUME 4 REPORT ON THE DEVELOPMENT AND HARMONISATION OF LAWS ON HAZARDOUS AND NON-HAZARDOUS WASTES 7$ ? 4 + December 1999 ISBN: 92-807-1887-8 PREFACE Environmental law is an essential tool for the governance East African countries are focused on issues which are and management of the environment and natural resources. essentially of sub-regional character. The management of the It is the foundation of national and regional policies and Joint Project is based at UNEP within its environmental law actions to ensure that the use of natural resources is done activities and is directed by a Task Manager, who works under equitably and sustainably. guidance of a Steering Committee. Members of the Steering Committee are UNEP, UNDP, FAO, The World Bank, IUCN In the East African sub-regional countries of Kenya, Tanzania Environmental Law Centre and The Dutch Government. and Uganda have, since 1995, been developing and harmoning various environmental laws in selected sectors THE PROCESS FOR DEVELOPMENT AND within their region. The process of developing and HARMONiZATION OF THE LAWS harmonizing environmental laws is intended to lead to the enactment or amendment of the internal legislative, regulatory Representatives of the three governments met in February and administrative framework of each country. Such change 1995 to work out general principles and modalities for their has been harmonized at a sub-regional level where the three cooperation. -

Open MYO THEIN-Phd Dissertation
The Pennsylvania State University The Graduate School Department of Engineering Science and Mechanics BIOCHIPS EMPLOYING MICROELECTRODES AND MICROTRANSDUCERS FOR CHARACTERIZATION AND PERMEABILIZATION OF CELL MEMBRANE AT THE WHOLE–CELL AND CELLULAR LEVEL A Dissertation in Engineering Science and Mechanics by Myo Min Thein © 2010 Myo Min Thein Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December 2010 The dissertation of Myo Min Thein was reviewed and approved* by the following: Jian Xu Associate Professor of Engineering Science and Mechanics and Adjunct Professor of Electrical Engineering Thesis Advisor and Chair of Committee Bernhard R. Tittmann Schell Professor of Engineering Science and Mechanics Yinong Yang Associate Professor of Plant Pathology Sulin Zhang Assistant Professor of Engineering Science and Mechanics Judith A. Todd P. B. Breneman Department Head Chair Head of the Department of Engineering Science and Mechanics * Signatures are on file in the Graduate School. ii Abstract In this study, two types of biochips, Integrated Microelectrode Array (IMA) chip and Ultrasonic Micro–Transducer Array (UMTA) biochip, were designed and micro– fabricated. The IMA chip employs cell–size microelectrode array architecture in order to electrically characterize cellular components, especially the properties of cell membrane, at the single–cell level. The UMTA biochip consists of arrays of high–aspect–ratio piezoelectric micro–transducers that are employed to efficiently permeabilize the cell membrane -

Velux Green Supply Chain Management for the Protection of the Environment and Human Health and Sustainable Development
VELUX RESTRICTED SUBSTANCE MANAGEMENT STANDARD VELUX GREEN SUPPLY CHAIN MANAGEMENT FOR THE PROTECTION OF THE ENVIRONMENT AND HUMAN HEALTH AND SUSTAINABLE DEVELOPMENT. VELUX RESTRICTED SUBSTANCES MANAGEMENT STANDARD (VRSMS) 1 September 2020 _____________________________________________________________________________________ INTRODUCTION For more than 75 years the VELUX Group (hereafter “VELUX”) has created better living environments by bringing daylight and fresh air into people's homes and commercial buildings all over the world. Our products help create bright, healthy, energy-efficient places in which to live, work, learn and play. VELUX has manufacturing and sales operations in more than 40 countries, with an extensive distribution network. Our products include a wide range of residential applications such as roof windows, skylights, flat roof windows and a wide range of commercial applications such as modular skylights with a wide range of installation solutions, roller shutters, blinds, decorative elements and remote controls. VELUX designs our products to minimise their impact on the environment and human health during sourcing, manufacture, use and disposal. To ensure that, we – as a downstream user - take a proactive and precautious approach to managing restricted substances and substances of high concern, VELUX has established a global chemical management process. Further, VELUX has established a management process for sustainable development. Our suppliers must be committed to support VRSMS generally and must comply with all environmental laws and regulations applicable for their business and must comply with VELUX internal restrictions in addition to the restrictions required by law or regulation (“VELUX Thresholds”) for the protection of the environment and human health as well as sustainable development. Further our suppliers must support us in our compliance with all environmental laws and regulations applicable for our business, including the issuance of compliance certificates and notification of first supply of substances. -

Guideline Ver6.2 Se Ria L No. CAS RN® Substance Group 1 Substance
Guideline Ver6.2 Rank CAS RN® Substance Group 1 Substance Group 2 Substance Name 1=Prohibited No. Serial 2=Reduction 1 124-73-2 ozone depleting substances(Halons) Ethane, 1,2-dibromo-1,1,2,2-tetrafluoro- 1 2 25497-30-7 ozone depleting substances(Halons) Dibromotetrafluoroethane 1 3 27336-23-8 ozone depleting substances(Halons) Dibromotetrafluoroethane,compressed 1 4 353-59-3 ozone depleting substances(Halons) Methane, bromochlorodifluoro- 1 5 354-56-3 ozone depleting substances(Halons) Ethane, pentachlorofluoro- 1 6 75-61-6 ozone depleting substances(Halons) Methane, dibromodifluoro- 1 7 75-63-8 ozone depleting substances(Halons) Methane, bromotrifluoro- 1 8 110003-27-5 ozone depleting substances(HBFCs) 1,3-dibromo-1,1,2-trifluoropropane 1 9 111483-20-6 ozone depleting substances(HBFCs) 1-bromo-2,3-difluoropropane 1 10 124-72-1 ozone depleting substances(HBFCs) 2-bromo-1,1,1,2-tetrafluoroethane 1 11 148875-95-0 ozone depleting substances(HBFCs) C3H3FBr4 1 12 148875-98-3 ozone depleting substances(HBFCs) C3H2F2Br4 1 13 1511-62-2 ozone depleting substances(HBFCs) Methane, bromodifluoro- 1 14 172912-75-3 ozone depleting substances(HBFCs) HBFC-131 B3 Tribromofluoroethane 1 15 1800-81-3 ozone depleting substances(HBFCs) 1-bromo-1,2-difluoroethane 1 16 1868-53-7 ozone depleting substances(HBFCs) Dibromofluoromethane 1 17 1871-72-3 ozone depleting substances(HBFCs) FLUOROBROMOPROPANE 1 18 2252-78-0 ozone depleting substances(HBFCs) 1-Bromo-1,1,2,3,3,3-hexafluoropropane 1 19 2252-79-1 ozone depleting substances(HBFCs) 2-bromo-1,1,1,3,3,3-hexafluoropropane -

Substances of Concern List
< KUBOTA Group Green Procurement Guidelines Appendix > April 2009 Established April 2010 Revised to Ver.2 April 2011 Revised to Ver.3 Substances of Concern List April 2011 KUBOTA Corporation Introduction This document is for providing information related to "3. Substances of Concern" of "Eco-friendliness standards for products" specified in "KUBOTA Group Green Procurement Guidelines" revised on April 2011. Table of Contents Number of Substances Table Number, Definition of Substances Page or Substance Groups Table 1 : Substances to be Prohibited Substances to be Prohibited means the substances which should not be 144 p3 contained in the products nor used in the production process. Table 2 : Substances to be Restricted Substances to be Restricted means the substances which should not be contained in the products nor used in the production process under the certain 7p6 conditions or applications. They are to be reduced gradually the content and use with the time limit and the target and to be promoted the substitution. Attached Table I-A : RoHS Exemptions List - p9 Attached Table I-B : ELV Exemptions List - p13 Table 3 : Substances to be Controlled Substances to be Controlled means the substances which should be recognized - p16 their presence in the products or use in the production process from the viewpoint of the environmental impact of the products life cycle. Reference List of Substances to be Prohibited, Restricted and Controlled Reference List of Substances to be Prohibited, Restricted and Controlled 4272 p17 exemplifies Substances to be Prohibited, Substances to be Restricted and Substances to be Controlled. Disclaimer While the information is made based on the related regulatory control etc. -

Potential Chemical Contaminants in the Marine Environment
Potential chemical contaminants in the marine environment An overview of main contaminant lists Victoria Tornero, Georg Hanke 2017 EUR 28925 EN This publication is a Technical report by the Joint Research Centre (JRC), the European Commission’s science and knowledge service. It aims to provide evidence-based scientific support to the European policymaking process. The scientific output expressed does not imply a policy position of the European Commission. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use that might be made of this publication. Contact information Name: Victoria Tornero Address: European Commission Joint Research Centre, Directorate D Sustainable Resources, Water and Marine Resources Unit, Via Enrico Fermi 2749, I-21027 Ispra (VA) Email: [email protected] Tel.: +39-0332-785984 JRC Science Hub https://ec.europa.eu/jrc JRC 108964 EUR 28925 EN PDF ISBN 978-92-79-77045-6 ISSN 1831-9424 doi:10.2760/337288 Luxembourg: Publications Office of the European Union, 2017 © European Union, 2017 The reuse of the document is authorised, provided the source is acknowledged and the original meaning or message of the texts are not distorted. The European Commission shall not be held liable for any consequences stemming from the reuse. How to cite this report: Tornero V, Hanke G. Potential chemical contaminants in the marine environment: An overview of main contaminant lists. ISBN 978-92-79-77045-6, EUR 28925, doi:10.2760/337288 All images © European Union 2017 Contents Acknowledgements ................................................................................................ 1 Abstract ............................................................................................................... 2 1 Introduction ...................................................................................................... 3 2 Compilation of substances of environmental concern ............................................. -

Front Matter
Edited by David T. Pierce and Julia Xiaojun Zhao Trace Analysis with Nanomaterials Related Titles Blackledge, R. D. (ed.) Hillenkamp, F., Peter-Katalinic, J. (eds.) Forensic Analysis on MALDI MS the Cutting Edge A Practical Guide to Instrumentation, New Methods for Trace Methods and Applications Evidence Analysis 2007 2007 ISBN: 978-3-527-31440-9 ISBN: 978-0-471-71644-0 Schalley, C. A. (ed.) Xu, X.-H. N. (ed.) Analytical Methods in New Frontiers in Supramolecular Chemistry Ultrasensitive Bioanalysis 2007 Advanced Analytical Chemistry ISBN: 978-3-527-31505-5 Applications in Nanobiotechnology, Single Molecule Detection, and Single Cell Analysis 2007 E-Book ISBN: 978-0-470-11949-5 Otto, M. Chemometrics Statistics and Computer Application in Analytical Chemistry 2007 ISBN: 978-3-527-31418-8 Edited by David T. Pierce and Julia Xiaojun Zhao Trace Analysis with Nanomaterials The Editors All books published by Wiley-VCH are carefully produced. Nevertheless, authors, editors, and Dr. David T. Pierce publisher do not warrant the information contained University of North Dakota in these books, including this book, to be free of Department of Chemistry errors. Readers are advised to keep in mind that 151 Cornell Street, Stop 9024 statements, data, illustrations, procedural details or Grand Forks, ND 58202-9024 other items may inadvertently be inaccurate. USA Library of Congress Card No.: applied for Dr. Julia Xiaojun Zhao University of North Dakota British Library Cataloguing-in-Publication Data Department of Chemistry A catalogue record for this book is available from 151 Cornell Street, Stop 9024 the British Library. Grand Forks, ND 58202-9024 USA Bibliographic information published by the Deutsche Nationalbibliothek The Deutsche Nationalbibliothek lists this publication in the Deutsche Nationalbibliografi e; detailed bibliographic data are available on the Internet at <http://dnb.d-nb.de>. -

Substances of Concern List
< KUBOTA Group Green Procurement Guidelines Appendix > Substances of Concern List January 2019 KUBOTA Corporation Introduction This document is for providing information related to "3. Substances of Concern" of "Eco-friendliness standards for products" specified in "KUBOTA Group Green Procurement Guidelines" revised on January 2019. Table of Contents Number of Substances Table Number, Definition of Substances Page or Substance Groups Table 1 : Substances to be Prohibited Substances to be Prohibited means the substances which should not be contained in the products 151 p3 nor used in the production process. Table 2 : Substances to be Restricted Substances to be Restricted means the substances which should not be contained in the products nor used in the production process under the certain conditions or applications. They are to be 19 p7 reduced gradually the content and use with the time limit and the target and to be promoted the substitution. Attached Table I-A : RoHS Exemptions List - p14 Table 3 : Substances to be Controlled Substances to be Controlled means the substances which should be recognized their presence in the - p21 products or use in the production process from the viewpoint of the environmental impact of the products life cycle. [Disclaimer] While the information is made based on the related regulatory control etc. of December 1, 2018, we make no warranties about the completeness and accuracy. Please confirm the original of the related regulatory control or the industry standards of the latest version properly in yourself when you use it. The publisher, Kubota Corporation, doesn't assume the responsibility of the damage suffers by using it. -
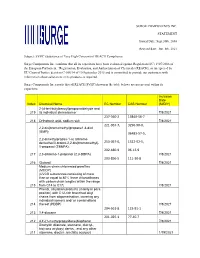
SURGE COMPONENTS INC. STATEMENT Issued Date: Sept.30Th, 2016 Revised Date: Jun. 8Th, 2021 Subject: SVHC (Substance of Very High
SURGE COMPONENTS INC. STATEMENT Issued Date: Sept.30th, 2016 Revised date: Jun. 8th, 2021 Subject: SVHC (Substance of Very High Concern) of REACH Compliance Surge Components Inc. confirms that all its capacitors have been evaluated against Regulation (EC) 1907/2006 of the European Parliament, “Registration, Evaluation, and Authorization of Chemicals (REACH), as interpreted by EU Court of Justice decision C-106/14 of 10 September 2015 and is committed to provide our customers with information about substances in its products as required. Surge Components Inc.reports that all REACH SVHC(shown in the table below) are not present within its capacitors. Inclusion Date Index Chemical Name EC Number CAS Number (M/D/Y) 2-(4-tert-butylbenzyl)propionaldehyde and 219 its individual stereoisomer 7/8/2021 237-560-2 13840-56-7 218 Orthoboric acid, sodium salt 7/8/2021 221-967-7, 3296-90-0, 2,2-bis(bromomethyl)propane1,3-diol (BMP); 36483-57-5, 2,2-dimethylpropan-1-ol, tribromo derivative/3-bromo-2,2-bis(bromomethyl)- 253-057-0, 1522-92-5, 1-propanol (TBNPA); 202-480-9 96-13-9 217 2,3-dibromo-1-propanol (2,3-DBPA) 7/8/2021 203-856-5 111-30-8 216 Glutaral 7/8/2021 Medium-chain chlorinated paraffins (MCCP) (UVCB substances consisting of more than or equal to 80% linear chloroalkanes with carbon chain lengths within the range 215 from C14 to C17) 7/8/2021 Phenol, alkylation products (mainly in para position) with C12-rich branched alkyl chains from oligomerisation, covering any individual isomers and/ or combinations 214 thereof (PDDP) 7/8/2021 204-661-8 123-91-1 213 1,4-dioxane 7/8/2021 201-025-1 77-40-7 212 4,4'-(1-methylpropylidene)bisphenol 7/8/2021 Dioctyltin dilaurate, stannane, dioctyl-, bis(coco acyloxy) derivs., and any other 211 stannane, dioctyl-, bis(fatty acyloxy) 1/19/2021 derivs. -

University of Cape Coast by George Yaw Hadzi October
UNIVERSITY OF CAPE COAST ASSESSMENT OF HEAVY METALS AND METALLOID IN SOILS, SEDIMENTS AND WATER FROM PRISTINE AND MAJOR MINING AREAS IN GHANA - DATA TO AID GEOCHEMICAL BASELINE . SETTING BY GEORGE YAW HADZI e Thesis submitted to the Department of Chemistry of the School of Physical Sciences, College of Agriculture and Natural Sciences, University of Cape Coast, in partial fulfilment of the requirements for the award of Doctor of Philosophy degree in Chemistry OCTOBER 2017 DECLARATION Candidate’s Declaration I hereby declare that this thesis is the result of my own original research and that no part of it has been presented for another degree in this university or elsewhere. Candidate’s Signature Date Name: GEORGE YAW HADZI Supervisors’ Declaration We hereby declare that the preparation and presentation of the thesis were supervised in accordance with the guidelines on supervision of thesis laid down by the University of Cape Coast Principal Supervisor’s Signature Dtae Name: PROF. DAVID KOFI ESSUMANG Co-Supervisor’s Signature ..Q? Name: PROF. SHILOH KWABENA DEDEH OSAE ii 4;^ JONAH LIBRARY OF CAPE COAST ABSTRACT The distribution, risks and geochemical baseline analysis of the heavy metals in soils, sediments and water in pristine and major mining areas in Ghana were investigated. The surface soils, sediment and water were pulverized, acid digested, and analyzed for the major and minor elements using (ICP- QQQMS). The average metals and metalloid (As, Cd, Pb and Zn) concentrations from the mining sites were higher than the pristine sites (p>0.05) with many of the metals not detected in pristine water samples. -

Flammable Chemicals 106 Ignition and Propagation of a Flame Front 106 Control Measures 147 Fire Extinguishment 149 Fire Precautions 151 Vi CONTENTS
Hazardous Chemicals Handbook P. A. Carson and C. J. Mumford Butterworth-Heinemann Ltd Linacre House, Jordan Hill, Oxford OX2 8DP -&A member of the Reed Elsevier group OXFORD LONDON BOSTON MUNICH NEW DELHl SINGAPORE SYDNEY TOKYO TORONTO WELLINGTON First published 1994 0 P. A. Carson and C. J. Mumford 1994 All rights reserved. No part of this publication may be reproduced in any material form (including photocopying or storing in any medium by electronic means and whether or not transiently or incidentally to some other use of this publication) without the written permission of the copyright holder except in accordance with the provisions of the Copyright. Designs and Patcnts Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd. 90 Tottenham Court Road, London, England W1P YHE. Applications for the copyright holder’s written permission to reproduce any part of this publication should be addressed to the publishers British Library Cataloguing in Publication Data Carson, P. A. Hazardous Chemicals Handbook 1. Title 11. Mumford, C. J. 363.17 ISBN 0 7506 0278 3 Library of Congress Cataloguing in Publication Data Carson, P. A., 1944- Hazardous chemicals handbook/P. A. Carson and C. J. Mumford. P. cm . Includes bibliographical references and index. ISBN 0 75(k 0278 3 1. Hazardous substances - Handbooks, manuals, etc. I. Mumford, C. J. 11. Title. T55.3.H3C368 93-7795 604.7-dc20 CIP Printed and bound in Great Britain Preface The aim of this handbook is to provide a source of rapid ready-reference to help in the often complex task of handling, using and disposing of chemicals safely and with minimum risk to people’s health or damage to facilities or to the environment.