Stereoselective Synthesis of Pyrrolidinones Via Nitro-Mannich Reaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Contemporary Organosilicon Chemistry
Contemporary organosilicon chemistry Edited by Steve Marsden Generated on 05 October 2021, 02:13 Imprint Beilstein Journal of Organic Chemistry www.bjoc.org ISSN 1860-5397 Email: [email protected] The Beilstein Journal of Organic Chemistry is published by the Beilstein-Institut zur Förderung der Chemischen Wissenschaften. This thematic issue, published in the Beilstein Beilstein-Institut zur Förderung der Journal of Organic Chemistry, is copyright the Chemischen Wissenschaften Beilstein-Institut zur Förderung der Chemischen Trakehner Straße 7–9 Wissenschaften. The copyright of the individual 60487 Frankfurt am Main articles in this document is the property of their Germany respective authors, subject to a Creative www.beilstein-institut.de Commons Attribution (CC-BY) license. Contemporary organosilicon chemistry Steve Marsden Editorial Open Access Address: Beilstein Journal of Organic Chemistry 2007, 3, No. 4. School of Chemistry, University of Leeds, Leeds LS2 9JT, UK doi:10.1186/1860-5397-3-4 Email: Received: 06 February 2007 Steve Marsden - [email protected] Accepted: 08 February 2007 Published: 08 February 2007 © 2007 Marsden; licensee Beilstein-Institut License and terms: see end of document. Abstract Editorial for the Thematic Series on Contemporary Organosilicon Chemistry. The field of organosilicon chemistry has a rich and varied the 1990s, and equivalent to the number appearing in the much history, and has long since made the progression from chemical longer established field of organoboron chemistry -

Methanesulfonyl Chloride
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

덴드리틱 벤질 클로라이드의 효율적인 합성 Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols W
Polymer(Korea), Vol. 31, No. 5, pp 417-421, 2007 덴드리틱 벤질 클로라이드의 효율적인 합성 이재욱F,7ㆍ한승철Fㆍ김희주ㆍ김정환ㆍ이언엽ㆍ김병기ㆍ성새름ㆍ강화신ㆍ김지현FFㆍ허도성FFF 동아대학교 화학과, F동아대학교 의생명과학과, FF동국대학교 생명화학공학과, FFF인제대학교 의생명화학과 (2007년 5월 9일 접수, 2007년 6월 29일 채택) Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols with Methanesulfonyl Chloride/Et3N Jae Wook Lee*,7, Seung Choul Han*, Hee Joo Kim, Jung Hwan Kim, Un Yup Lee, Byoung-Ki Kim, Sae Reum Sung, Hwa-Shin Kang, Ji Hyeon Kim**, and Do-Sung Huh*** Department of Chemistry, Dong-A University, Hadan-2-dong, Busan 604-714, Korea *Department of Medical Bioscience, Dong-A University, Hadan-2-dong, Busan 604-714, Korea **Department of Chemical & Biochemical Engineering, Dongguk University, Seoul 100-715, Korea ***Department of Biomedicinal Chemistry, Inje University, Gim Hai 621-749, Korea (Received May 9, 2007; Accepted June 29, 2007) 초록: 덴드리틱 벤질 알코올을 트리에틸아민과 메탄술포닐클로라이드와 반응시켜서, 덴드리틱 벤질 클로라이드 의 효율적인 합성이 이루어졌다. 이 반응은 히드록시기의 메실화 반응과 염소화 반응의 2단계 반응으로 이루어지 는데, 중간체의 분리없이 한 반응 용기내에서 반응이 진행되는 경제적인 방법이다. Abstract: A successful rapid synthesis of dendritic benzyl chlorides from dendritic benzyl alcohols using methanesulfonyl chloride/Et3N as activating agents was described. In this method, each dendritic benzyl chloride can be prepared in one pot: no isolation of intermediate mesylated dendrons is required. The key steps in the syntheses of dendritic benzyl chlorides were the mesylation of the hydroxymethyl group followed by the chlorination by in-situ generated triethylammonium chloride. Keywords: benzylic alcohol, chlorination. Introduction vation and coupling steps in high yield. In addition, the coupling step must be very efficient to enable complete Dendrimers represent a novel type of polymeric material reaction. -
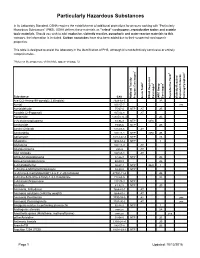
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

Fragment-Based Discovery of a New Class of Inhibitors Targeting Mycobacterial Trna Modification
Supplementary Data Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification Sherine E. Thomas*1, Andrew J. Whitehouse*2, Karen Brown3,4, Juan M. Belardinelli5, Ramanuj Lahiri6, M. Daben J. Libardo7, Pooja Gupta1, Sony Malhotra8, Helena I. M. Boshoff7, Mary Jackson5, Chris Abell 2, Anthony G. Coyne2, Tom L. Blundell §1, R. Andres Floto3,4, Vitor Mendes §1 1 Department of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge CB2 1GA, UK. 2 Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. 3 MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge, CB2 0QH, U.K. 4 Cambridge Centre for Lung Infection, Royal Papworth Hospital, Cambridge, CB23 3RE, U.K. 5 Mycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, Colorado, USA. 6 National Hansen’s Disease Program, Healthcare Systems Bureau, HRSA, DHHS, USA 7 Tuberculosis Research Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Disease, National Institutes of Health, 9000 Rockville Pike, Bethesda, Maryland 20892, USA. 8 Birkbeck College, University of London, Malet Street, WC1E7HX, UK * Contributed equally § To whom correspondence should be addressed Results Conservation of catalytic residues in mycobacterial TrmD A multiple sequence alignment of 5 mycobacterial TrmD enzyme sequences with 10 TrmD ortholog amino acid sequences (Figure S1) shows conservation of important catalytic residues and residues involved in tRNA recognition and methyl transfer reactions. The catalytic residues Asp169 and Arg154 are highly conserved across TrmD orthologs. Key residues such as Asp50, Gly59, His46 involved in tRNA G36 and G37 base recognition, interactions with bases at positions 38, 32 and anticodon branch of wild type tRNA respectively, are also broadly conserved. -

4 Methanesulfonyl Chloride1 Acute Exposure Guideline Levels
Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies THE NATIONAL ACADEMIES PRESS 500 FIFTH STREET, NW WASHINGTON, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Insti- tute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was supported by Contract No. W81K04-11-D-0017 and EP-W-09-007 be- tween the National Academy of Sciences and the U.S. Department of Defense and the U.S. Environmental Protection Agency. Any opinions, findings, conclusions, or recom- mendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the organizations or agencies that provided support for this project. International Standard Book Number-13: 978-0-309-28308-3 International Standard Book Number-10: 0-309-28308-6 Additional copies of this report are available for sale from the National Academies Press, 500 Fifth Street, NW, Keck 360, Washington, DC 20001; (800) 624-6242 or (202) 334- 3313; http://www.nap.edu/. Copyright 2013 by the National Academy of Sciences. All rights reserved. Printed in the United States of America The National Academy of Sciences is a private, nonprofit, self-perpetuating society of distinguished scholars engaged in scientific and engineering research, dedicated to the furtherance of science and technology and to their use for the general welfare. -

Guide for the Selection of Chemical and Biological Decontamination Equipment for Emergency First Responders
U.S. Department of Justice Office of Justice Programs National Institute of Justice National Institute of Justice Law Enforcement and Corrections Standards and Testing Program Guide for the Selection of Chemical and Biological Decontamination Equipment for Emergency First Responders NIJ Guide 103–00 Volume I October 2001 ABOUT THE LAW ENFORCEMENT AND CORRECTIONS STANDARDS AND TESTING PROGRAM The Law Enforcement and Corrections Standards and Testing Program is sponsored by the Office of Science and Technology of the National Institute of Justice (NIJ), U.S. Department of Justice. The program responds to the mandate of the Justice System Improvement Act of 1979, which directed NIJ to encourage research and development to improve the criminal justice system and to disseminate the results to Federal, State, and local agencies. The Law Enforcement and Corrections Standards and Testing Program is an applied research effort that determines the technological needs of justice system agencies, sets minimum performance standards for specific devices, tests commercially available equipment against those standards, and disseminates the standards and the test results to criminal justice agencies nationally and internationally. The program operates through: The Law Enforcement and Corrections Technology Advisory Council (LECTAC), consisting of nationally recognized criminal justice practitioners from Federal, State, and local agencies, which assesses technological needs and sets priorities for research programs and items to be evaluated and tested. The Office of Law Enforcement Standards (OLES) at the National Institute of Standards and Technology, which develops voluntary national performance standards for compliance testing to ensure that individual items of equipment are suitable for use by criminal justice agencies. -

Review 0103 - 5053 $6.00+0.00
http://dx.doi.org/10.5935/0103-5053.20150045 J. Braz. Chem. Soc., Vol. 26, No. 5, 837-850, 2015. Printed in Brazil - ©2015 Sociedade Brasileira de Química Review 0103 - 5053 $6.00+0.00 Recent Syntheses of Frog Alkaloid Epibatidine Ronaldo E. de Oliveira Filho and Alvaro T. Omori* Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, 09210-580 Santo André-SP, Brazil Many natives from Amazon use poison secreted by the skin of some colorful frogs (Dendrobatidae) on the tips of their arrows to hunt. This habit has generated interest in the isolation of these toxins. Among the over 500 isolated alkaloids, the most important is undoubtedly (-)-epibatidine. First isolated in 1992, by Daly from Epipedobates tricolor, this compound is highly toxic (LD50 about 0.4 µg per mouse). Most remarkably, its non-opioid analgesic activity was found to be about 200 times stronger than morphine. Due to its scarcity, the limited availability of natural sources, and its intriguing biological activity, more than 100 synthetic routes have been developed since the epibatidine structure was assigned. This review presents the recent formal and total syntheses of epibatidine since the excellent review published in 2002 by Olivo et al.1 Mainly, this review is summarized by the method used to obtain the azabicyclic core. Keywords: epibatidine, organic synthesis, azanorbornanes H Cl H 1. Introduction N N O N N At an expedition to Western Ecuador in 1974, Daly and Myers isolated traces of an alkaloid with potential biological (–)-Epibatidine(1) Epiboxidine(1a) activity from the skin of the species Epipedobastes tricolor. -

Chem. Pharm. Bull. 68(1): 1-33 (2020)
Vol. 68, No. 1 Chem. Pharm. Bull. 68, 1–33 (2020) 1 Review Development of New Synthetic Reactions Using Hetero-Heavy Atoms and Their Application to Synthesis of Biofunctional Molecules Shinya Harusawa Osaka University of Pharmaceutical Sciences; 4–20–1 Nasahara, Takatsuki, Osaka 569–1094, Japan. Received July 30, 2019 Novel reactions using hetero-heavy atoms (P, S, Si, Se, and Sn) were developed and applied to the syn- thesis of biofunctional molecules and some medicine-candidates, in which the following items are covered. 1) Development of introduction of C1-unit using cyanophosphates (CPs). 2) Carbene-generation under neutral condition from CPs and its application to organic synthesis. 3) [3,3]Sigmatropic rearrangement-ring expan- sion reactions of medium-sized cyclic thionocarbonates containing a sulfur atom and their application to natural product synthesis. 4) Stereoselective synthesis of novel β-imidazole C-nucleosides via diazafulvene intermediates and their application to investigating ribozyme reaction mechanism. 5) Developments of novel histamine H3- and H4-receptor ligands using new synthetic methods involving Se or Sn atoms. Key words cyanophosphate; carbene; sigmatropic rearrangement; ribozyme; histamine H3 receptor; antagonist 1. Introduction with DEPC to afford α-amino nitriles, the function of which is Organic synthesis has reached a high degree of perfection. easily converted into α-amino acids2) (Chart 2, Eq. 1); modi- However, reactions occurring within a living organism are fied Strecker synthesis for the preparation of α-amino nitriles much more mild, and more highly efficient, with specific- using DEPC from carbonyl compounds and amines in tetra- ity and selectivity. Thus, new efficient synthetic methods and hydrofuran (THF) under mild reaction conditions (Eq. -

Bio-Organic Mechanism Game – Simplistic Biochemical Structures and Simplistic Organic Reaction Mechanisms Are Used to Explain Common Biochemical Transformations
1 Bio-Organic Mechanism Game – Simplistic biochemical structures and simplistic organic reaction mechanisms are used to explain common biochemical transformations. Simplified biochemical molecules are presented first. Many biomolecules have a somewhat complex structure that makes it difficult to write out step by step mechanisms. However, if we simplify those structures to the essential parts necessary to explain the mechanistic chemistry of each step, it becomes much easier to consider each step through an important cycle. I have proposed possible simplified structures that are used in the later examples of biochem cycles and problems. The usual strategy in biochem cycles is to just write names, or perhaps, names and a structure. Occasionally a few mechanistic steps are suggested, but almost never is a detailed sequence of mechanistic steps provided. Since it is hard to find such detailed mechanistic steps anywhere (sometimes they are not known) our proposed steps are, of necessity, somewhat speculative. In this book we are not looking for perfection, which is not possible, but for sound organic logic that is consistent with the biochemical examples presented below. There is great satisfaction in blending organic knowledge with real life reactions that help explain how life works. In working through some of the problems, you may develop an alternative mechanism that is just as good, or even better than the one I have proposed. If you do, I hope you will share it with me and if an improved version of this book ever gets written I can include it the next edition (and give you credit). It is almost certain that I have made some errors and I would appreciate it if you would let me know about them. -

Chapter 29 Information
Chapter 29 Information There are three types of organic reactions. 1. Polar reactions. These are of the SN1/SN2/EAS/etc. type; a nucleophile reacts with an electrophile, and both electrons in the new bond come from the nucleophile. 2. Radical reactions. A new bond is formed using one electron from each of the reactants. 3. Pericyclic reactions. The electrons in one or more reactants are reorganized in a cyclic manner. There are three major types. a. Electrocyclic. An intramolecular reaction in which a new sigma bond is formed between the ends of a conjugated pi system. The product is cyclic, with one more ring and one less pi bond than the starting material. These reactions are reversible. b. Cycloaddition. Two different molecules, each containing at least one pi bond, react to form a cyclic compound. Each loses a pi bond, and two new sigma bonds are formed. The Diels‐Alder reaction is a classic cycloaddition. c. Sigmatropic rearrangement. A sigma bond is broken, a new sigma bond is formed, and the pi bonds rearrange. Features of pericyclic reactions include: • They are concerted reactions. Therefore there is one transition state and no intermediate. • They are highly stereoselective (because they are concerted). • They are generally not affected by catalysts or by changes in the solvent. The configuration of the product is determined by: • The configuration of the starting material(s). • The number of pi electrons (whether conjugated pi bonds or lone pairs) in the reacting system. • Whether the reaction is done under thermal or photochemical conditions. o Photochemical: when the reactant absorbs light (usually indicated with hν).