Synthesis and Reactivity of Cyclopropanes and Cyclopropenes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Docteur» at the University François Rabela
UNIVERSITÉ FRANÇOIS – RABELAIS DE TOURS École Doctorale « Santé - Sciences Biologiques - Chimie du Vivant » and UNIVERSITY OF LJUBLJANA, FACULTY OF PHARMACY «Department of Pharmaceutical Chemistry» A cotutelle thesis submitted in fulfillment of the requirements for the degree of «Docteur» at the University François Rabelais of Tours (France) and Doctor of Pharmacy at the University of Ljubljana (Slovenia) In Pharmaceutical Chemistry Publicly defended on the 1st of March 2013 by Mitja KOVAČ in Ljubljana FLUORATION DE DERIVES DU BENZOVESAMICOL POUR L'OBTENTION DE RADIOLIGANDS POTENTIELS DU TRANSPORTEUR VESICULAIRE DE L'ACETYLCHOLINE Under the co-direction of: Associate Professor Sylvie Mavel (MCU, Tours) and Associate Professor Marko Anderluh (Ljubljana) ----------------- JURY for Oral Defense: Ms MAVEL Sylvie – Associate Professor, University François-Rabelais, Tours, France Mr ANDERLUH Marko – Associate Professor, University of Ljubljana, Slovenia Mr DOLLÉ Frédéric – Service Hospitalier Frédéric Joliot, Institut d'Imagerie BioMédicale - CEA, Orsay, France (Reviewer) Mr EMOND Patrick – Professor, University François-Rabelais, Tours, France Ms GMEINER STOPAR Tanja – Assistant Professor, University of Ljubljana, Slovenia (Reviewer) Mr GOBEC Stanislav – Professor, University of Ljubljana, Slovenia (Chairman) This cotutelle PhD was carried out with the collaboration between the University of Tours (Laboratoire de Biophysique Médicale et Pharmaceutique, Unité INSERM U930 - FRANCE) and the University of Ljubljana (Faculty of Pharmacy, Department of Pharmacutical Chemistry - SLOVENIA). The work was supported by a grant from the Slovene Human Resources Development and Scholarship Fund, by a grant from the University of Ljubljana (Inovativna shema za sofinanciranje doktorskega študija za spodbujanje sodelovanja z gospodarstvom in reševanja aktualnih družbenih izzivov - generacija 2010 Univerza v Ljubljani), and by a Slovenia- French bilateral collaboration project (project n° BI-FR/12-13-PROTEUS-007). -

Friction of Iron Lubricated with Aliphatic and Aromatic Hydrocarbons and Halogenated Analogs
NASA TECHNICAL NOTE 00 0 N w n a c 4 &A 4 a w~.~,z..p~COPY: RETURN TO --;F?.f-..wL TECHNICAL LIBRARY 1(LWTWND ATS, M* M* FRICTION OF IRON LUBRICATED WITH ALIPHATIC AND AROMATIC HYDROCARBONS AND HALOGENATED ANALOGS Donald H. Buckley Lewis Research Center NATIONAL AERONAUTICS AND SPACE ADMINISTRATION WASHINGTON, D. C. APRIL 1976 TECH LIBRARY KAFB,"I .-- - -.- ~~ OL337b7 I 1. Report No. I 2. Government Accession No. ]Recipient's Catalog NO. TN D -8208 I- .- .. I I 4. Title and Subtitle 5. Report Date FRICTION OF IRON LUBRICATED WITH ALIPHATIC AND I April 1976 Performing OrganizationCode AROMATIC HYDROCARBONS AND HALOGENATED ANALOGS I 7. Author(s1 8. Performing Organization Report No. E-8558 Donald H. Buckley ~__ .. 10. Wcrk Unit No. 9. Performing Organization Name and Address 506-16 Lewis Research Center 11. Contract or Grant No. National Aeronautics and Space Administration I Cleveland, Ohio 44135 13. Type of Report and Period Covered ~~ 12. Sponsoring Agency Name and Address Technical Note National Aeronautics and Space Administration 14. Sponsoring Agency Code Washington, D.C. 20546 I 1 15. Supplementary Notes - -- L16. Abstract An investigation was conducted to determine the influence of oxygen and various organic mole cules on the reduction of the friction of an iron (011) single crystal surface. A comparison was made between aliphatic and aromatic structures, all of which contained six carbon atoms, and among various halogen atoms. Results of the investigation indicate that hexane and benzene give similar friction coefficients over a range of loads except at very light loads. At light loads, the friction decreased with an increase in the load where the halogens fluorine and chlorine are in corporated into the benzene molecular structure; however, over the same load range when bro mine and iodine were present, the friction was relatively unchanged. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -
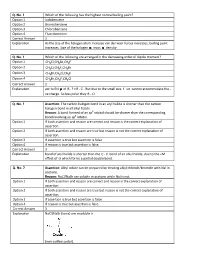
Q. No. 3 Which of the Following Has the Highest Normal Boiling Point
Q. No. 3 Which of the following has the highest normal boiling point? Option 1 Iodobenzene Option 2 Bromobenzene Option 3 Chlorobenzene Option 4 Fluorobenzene Correct Answer 1 Explanation As the size of the halogen atom increase van der waal forces increases, boiling point increases. Size of the halogen mass density. Q. No. 4 Which of the following are arranged in the decreasing order of dipole moment? Option 1 CH3 Cl,CH 3 Br,CH 3 F Option 2 CH3 Cl,CH 3 F,CH 3 Br Option 3 CH3 Br,CH 3 Cl,CH 3 F Option 4 CH3 Br,CH 3 F,CH 3 Cl Correct Answer 2 Explanation acc to EN of RF> RCl . But due to the small size. F ion cannot accommodate the ve charge. So less polar they R Cl . Q. No. 5 Assertion : The carbon halogen bond in an aryl halid e is shorter than the carbon halogen bond in all alkyl halide. Reason : A bond formed of an sp 3 orbital should be shorter than the corresponding bond involving an sp 2 orbital. Option 1 If both assertion and reason are correct and reason is the correct explanation of assertion. Option 2 If both assertion and reason are true but reason is not the correct explanation of assertion. Option 3 If assertion is true but assertion is false. Option 4 If reason is true but assertion is false. Correct Answer 3 Explanation bond of aryl halide is shorter than the C X bond of an alkyl halide, due to the +M effect of X which forms a partial double bond. -

Acenaphthene 83-32-9 5000 N 45000 N 100000 L 110 N 530 N Acephate 30560-19-1 110 N 980 N 2100 N 0.11 N 24 N Acetaldehyde 75-07-0
Table A-6: 2021 Screening Levels Soil Exposure Ground Water Vapor Exposure Chemical Direct Contact Soil MTG Tap Ground Water Indoor Air Residential Com/Ind Excavation Residential Residential Residential Com/Industrial Residential Com/Ind Name CASRN (mg/kg) (mg/kg) (mg/kg) (mg/kg) (ug/L) (ug/L) (ug/L) (ug/m3) (ug/m3) Acenaphthene 83-32-9 5000 N 45000 N 100000 L 110 N 530 N Acephate 30560-19-1 110 N 980 N 2100 N 0.11 N 24 N Acetaldehyde 75-07-0 110 N 340 N 1900 N 0.077 N 19 N 9.4 N 39 N Acetochlor 34256-82-1 1800 N 16000 N 34000 N 5.6 N 350 N Acetone 67-64-1 85000 N 100000 L 100000 L 57 N 14000 N 32000 N 140000 N Acetone Cyanohydrin 75-86-5 100000 L 100000 L 100000 L 2.1 N 8.8 N Acetonitrile 75-05-8 1100 N 3400 N 19000 N 0.54 N 130 N 63 N 260 N Acetophenone 98-86-2 2500 S 2500 S 2500 S 12 N 1900 N Acetylaminofluorene, 2- 53-96-3 2 C 6 C 320 C 0.015 C 0.16 C 0.022 C 0.094 C Acrolein 107-02-8 0.2 N 0.6 N 3.4 N 0.00017 N 0.042 N 0.021 N 0.088 N Acrylamide 79-06-1 3.4 C 46 C 2400 C 0.0021 C 0.5 C 0.1 C 1.2 C Acrylic Acid 79-10-7 140 N 420 N 2300 N 0.0085 N 2.1 N 1 N 4.4 N Acrylonitrile 107-13-1 3.5 C 11 C 370 N 0.0023 C 0.52 C 0.41 C 1.8 C Adiponitrile 111-69-3 100000 L 100000 L 100000 L 6.3 N 26 N Alachlor 15972-60-8 140 C 410 C 18000 N 0.033 M 2 M Aldicarb 116-06-3 88 N 820 N 1800 N 0.015 M 3 M Aldicarb Sulfone 1646-88-4 88 N 820 N 1800 N 0.0088 M 2 M Aldicarb sulfoxide 1646-87-3 0.018 M 4 M Aldrin 309-00-2 0.55 C 1.8 C 59 N 0.03 C 0.0092 C 0.0057 C 0.025 C Allyl Alcohol 107-18-6 4.9 N 15 N 83 N 0.00086 N 0.21 N 0.1 N 0.44 N Allyl Chloride -

Process for Preparing Fluorobenzene
European Patent Office 0 Publication number: 0 054 274 Office europeen des brevets A1 tUnUPhAN PATENT APPLICATION (21J Application number: 81110361.3 © Int. CI.3: C 07 C 25/13 C 07 C 17/28 © Dateoffiling: 11.12.81 \m) Priority: 13.1Z.80 JP 176132/80 72) Inventor: Shimokawa, Kazuhiro 28.12.80 JP 186638/80 48-2, Suito-cho Suita-shi Osaka(JP) (43) Date of publication of application: 72) Inventor: Naito, Daiji 23.06.82 Bulletin 82/25 21-6, Takada-cho Ibaraki-shi Osaka(JP) @ Designated Contracting States: DE FR GB §) Inventor: Misaki, Susumu 240-141, Oaza-Aomadani Mino-shi @ Applicant: Daikin Kogyo Co., Ltd. Osaka(JP) No 1-12-39, Umeda Kita-ku Osaka-shi Osaka-fu(JP) Inventor: Yoshida, Tsutomu 2-21-21, Hitotsuya Settsu-shi @ Inventor: Tabushi, Iwao Osaka(JP) 24, Higashisakuragi-cho Matsugasaki Sakyo-ku Kyoto Kyoto(JP) 75) Representative: von Kreisler, Alek, Dipl.-Chem. et al, Deichmannhaus am Hauptbahnhof D-5000 Kolnl(DE) 54) Process for preparing fluorobenzene. Monofluorobenzene is prepared in a good yield by reacting cyclopentadiene or its dimer of the formula: with fluorohalomethane of the formula: wherein Xs are, same or different, halogen atoms in the presence of a phase transfer catalyst and an acid acceptor. This invention relates to a process for preparing fluorobenzene. More particularly, it relates to a process for preparing fluorobenzene by reacting cyclopentadiene or its dimer, bicyclopentadiene, with fluorohalomethane. Fluorobenzene is a very useful compound as a starting material in preparation of medicines, agricultural chemicals, -

Composite Worker Air RSL May 2021 HQ01
Regional Screening Level (RSL) Composite Worker Ambient Air Table (TR=1E-06, HQ=0.1) May 2021 Key: I = IRIS; P = PPRTV; O = OPP; A = ATSDR; C = Cal EPA; X = PPRTV Screening Level; H = HEAST; W = TEF applied; E = RPF applied; G = user's guide Section 5; M = mutagen; V = volatile; R = RBA applied ; c = cancer; n = noncancer; * = where: n SL < 100X c SL; ** = where n SL < 10X c SL; SSL values are based on DAF=1; m = ceiling limit exceeded; s = Csat exceeded. Noncancer Hazard Index (HI) = Toxicity and Chemical-specific Information Contaminant Carcinogenic Target Risk (TR) = 1E-06 0.1 k k v Carcinogenic SL Noncarcinogenic SL IUR e RfCi e o TR=1E-06 THI=0.1 (ug/m3)-1 y (mg/m3) y l mutagen Analyte CAS No. (ug/m3) (ug or fibers/m3) Acephate 30560-19-1 2.2E-06 I 9.0E-03 I V Acetaldehyde 75-07-0 5.6E+00 3.9E+00 Acetochlor 34256-82-1 3.1E+01 A V Acetone 67-64-1 1.4E+04 2.0E-03 X Acetone Cyanohydrin 75-86-5 8.8E-01 6.0E-02 I V Acetonitrile 75-05-8 2.6E+01 V Acetophenone 98-86-2 1.3E-03 C Acetylaminofluorene, 2- 53-96-3 9.4E-03 2.0E-05 I V Acrolein 107-02-8 8.8E-03 1.0E-04 I 6.0E-03 I M Acrylamide 79-06-1 1.2E-01 2.6E+00 1.0E-03 I V Acrylic Acid 79-10-7 4.4E-01 6.8E-05 I 2.0E-03 I V Acrylonitrile 107-13-1 1.8E-01 8.8E-01 6.0E-03 P Adiponitrile 111-69-3 2.6E+00 Alachlor 15972-60-8 Aldicarb 116-06-3 Aldicarb Sulfone 1646-88-4 Aldicarb sulfoxide 1646-87-3 4.9E-03 I V Aldrin 309-00-2 2.5E-03 1.0E-04 X V Allyl Alcohol 107-18-6 4.4E-02 6.0E-06 C 1.0E-03 I V Allyl Chloride 107-05-1 2.0E+00 4.4E-01 5.0E-03 P Aluminum 7429-90-5 2.2E+00 Aluminum Phosphide 20859-73-8 -

Fluorobenzene and Chlorobenzene Are More Reactive Toward the Resulting +S02CH3 Than the Unshared Elec
6221 Table I. Nmr Parameters of Alkylarylhalonium Ions= Ion CH,X+- CHI -CHI- CH3CX+ Aromatic 19Fb C&-Br+-CH3 4.45 7.5-7.9 C&I&-I+-CHa 3.80 7.7-8.3 4-FCeH4-I+-CH3 3.80 7.0-8.1 103.4 4-FC6H4-Br+-CH3 4.40 7.3-8.1 103.7 4-CH3C&14-If-CH3 3.75 2.40 7.3-8.0 4-CH8C&14-Br+-CH3 4.35 2.40 7.4-7.9 C6HS-I+-C2H5 4.70 1.95 7.6-8.1 4-FCeH 4-I+-C2H 5 4.75 1.92 7.2-8,2 104.2 Cd-15-Br+-C2H5 5.30 1.90 7.7-8,2 4-FC sH 4-Br+-C~H 5.35 I .90 7.3-8.1 104.1 a Proton chemical shifts are from TMS in external capillary tube. Spectra were recorded at -70" in SO2 solution at 60 MHz. Relative peak areas were in agreement with expectation. Fluorine chemical shifts are from CFBCCI.Iin external capillary tube. The chemical shifts of 4-fluorobromobenzene and 4-fluoroiodobenzene in SO1 solution are 114.8 and 113.9 ppm, respectively. fluorobenzene and chlorobenzene are more reactive The fact that one gets increasing amounts of the ortho toward the resulting +S02CH3than the unshared elec- alkylated isomer going through the series from fluoro- tron pairs on halogen. The fluorine atom in any case benzene to iodobenzene in Friedel-Crafts a1 kylations is too electronegative to form a fluoronium ion. When has been generally explained by an intramolecular chlorobenzene is added to methyl fluoroantimonate in rearrangement of the intermediate alkylarylhalonium SO2C1F, sulfinylmethylation does not occur while ions. -

Development of Rules of Attraction for Intercalated Guest Molecules Inside of a Hydrogen Bonded Framework Matthew If Scher [email protected]
University of Missouri, St. Louis IRL @ UMSL Dissertations UMSL Graduate Works 4-20-2018 Development of Rules of Attraction for Intercalated Guest Molecules Inside of a Hydrogen Bonded Framework Matthew iF scher [email protected] Follow this and additional works at: https://irl.umsl.edu/dissertation Part of the Analytical Chemistry Commons, Inorganic Chemistry Commons, and the Materials Chemistry Commons Recommended Citation Fischer, Matthew, "Development of Rules of Attraction for Intercalated Guest Molecules Inside of a Hydrogen Bonded Framework" (2018). Dissertations. 731. https://irl.umsl.edu/dissertation/731 This Dissertation is brought to you for free and open access by the UMSL Graduate Works at IRL @ UMSL. It has been accepted for inclusion in Dissertations by an authorized administrator of IRL @ UMSL. For more information, please contact [email protected]. Development of Rules of Attraction for Intercalated Guest Molecules Inside of a Hydrogen Bonded Framework. By Matthew Joseph Fischer, Sr M.Sc., Chemistry, Saint Louis University, 2006 B.A. Chemistry, Saint Louis University, 2003 A DISSERTATION Submitted to The Graduate School at the UNIVERSITY OF MISSOURI – ST. LOUIS In Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY in CHEMISTRY with an emphasis in Inorganic Chemistry May 2018 Advisory Committee Alicia M. Beatty, Ph.D. Chairperson George Gokel, Ph.D. Stephen Holmes, PhD James Chickos, PhD 1 Abstract Development of Rules of Attraction for Intercalated Guest Molecules Inside of a Hydrogen Bonded Framework. (May 2018) Matthew Joseph Fischer, Sr M.Sc., Chemistry, Saint Louis University, 2006 B.A. Chemistry, Saint Louis University, 2003 Chair of Committee: Dr. Alicia M. -
Dissertation
DISSERTATION Titel der Dissertation „Synthesis of Novel PET-Tracers for the Norepinephrine Transporter“ verfasst von Christina Rami-Mark, Bakk. MSc. angestrebter akademischer Grad Doktorin der Naturwissenschaften (Dr. rer. nat.) Wien, 2015 Studienkennzahl lt. Studienblatt: A 796 605 419 Dissertationsgebiet lt. Studienblatt: Chemie Betreut von: Assoc. Prof. Mag. Dr. Wolfgang Wadsak 2 “Discovery consists of looking at the same thing as everyone else and thinking something different.” ― Albert Szent-Györgyi 3 4 ACKNOWLEDGEMENTS "You don't write because you want to say something; you write because you've got something to say." -F. Scott Fitzgerald Most of all, I would like to thank my supervisor and mentor Prof. Dr. Wolfgang Wadsak for his endless support, understanding, guidance and discussions. He gave me the possibility to take my own path, to learn a lot and has placed his trust on me all the way. Danke Wolfgang, dass du mich immer unterstützt hast, danke für deinen Rat und Hilfe, wenn mal was nicht so funktioniert hat und für dein Vertrauen in mich. I would also like to express my gratitude to Prof. Dr. Markus Mitterhauser for his support and bracing discussions. Danke Markus, für all die inspirierenden Meetings und deine Unterstützung. Thanks to Dr. Johanna Obermair, she made me learning a lot. Danke Jo, für alles was du mir beigebracht hast. To all my teammates and friends at the Radiochemistry and Biomarker Development unit, helping me survive all this stress in the orange world of the AKH by always cheering me up. Thanks for your patience when knocking on your doors asking for help. -

Alksli and Then Tested Qualitatively for Fluorine by a Potassium Fusion Downloaded by Guest on October 2, 2021 184 CHEMISTRY: BANCROFT and WHEARTY, JR
VOL. 17, 1931 CHEMISTRY: BANCROFT AND WHEARTY, JR. 183 1. Purified, activated charcoal causes chlorine to substitute in the ring with benzene and toluene. 2. We have confirmed Custis' results that light from an iron arc causes some ring substitution of chlorine with benzene and with toluene. 3. Certain unspecified wave-lengths in the ultra-violet activate benzene and toluene and give rise to the ring substitution. 4. If the action of the charcoal is not due to metallic impurities, char- coal must be an activating catalyst for benzene and toluene. 1 This work is part of the programme now being carried out at Cornell University under a grant from the Heckscher Foundation for the Advancement of Research estab- lished by August Heckscher at Cornell University. 2 Bancroft, J. Phys. Chem., 12, 417 (1908). 3 Willgerodt, J. prakt. Chem., [2] 34, 264 (1886). Slator, Z. physik. Chem., 45, 513 (1903). Goldberg, Z. wiss. Photographie, 4, 61 (1906). 6 Schramm, Ber., 18, 606 (1885). 7 Cannizzaro, Compt. rend., 41, 517 (1855). 8 Colson and Gautier, Ann. chim. phys., [6] 11, 19 (1887). Custis, J. Franklin Inst., 184, 875 (1917). 10 Bornwater and Holleman, Rec. trav. chim., 32, 231 (1913). AROMA TIC SUBSTITUTION PROD UCTS WITH FL UORINE' By WILDER D. BANCROFT AND S. F. WHEARTY, JR. BAKER CHEMICAL LABORATORY, CORNELL UNIVERSITY Communicated February 24, 1931 When fluorine is passed into dry benzene at room temperature, small explosions occur and carbon is precipitated, hydrogen fluoride and carbon tetrafluoride passing off. If the fluorine is diluted to one-half with dry nitrogen, and the benzene is cooled to 70, the reaction takes place more slowly and a tarry product is obtained with an odor somewhat like di- phenyl. -

Electrochemical Investigation of Homogenous Species at an Electrode Interface Christopher Larsen III
University of New Mexico UNM Digital Repository Chemistry ETDs Electronic Theses and Dissertations 6-24-2015 Electrochemical Investigation of Homogenous Species at an Electrode Interface Christopher Larsen III Follow this and additional works at: https://digitalrepository.unm.edu/chem_etds Part of the Physical Chemistry Commons Recommended Citation Larsen, Christopher III. "Electrochemical Investigation of Homogenous Species at an Electrode Interface." (2015). https://digitalrepository.unm.edu/chem_etds/41 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Chemistry ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Christopher A. Larsen III Candidate Chemistry & Chemical Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Richard Kemp , Chairperson Martin Kirk Ramesh Giri Zachary Sharp i Electrochemical Investigation of Homogenous Species at an Electrode Interface BY CHRISTOPHER ALAN LARSEN III Bachelor of Science in Chemistry University of New Mexico 2010 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Chemistry The University of New Mexico Albuquerque, New Mexico April 6, 2015 ii “Misfortune is needed to plumb certain mysterious depths in the understanding of men; pressure is needed to explode the charge. My captivity concentrated all my faculties on a single point. They had previously been dispersed, now they clashed in a narrow space; and, as you know, the clash of clouds produces electricity, electricity produces lightning and lightning gives light.” The Count of Monte Cristo, Alexander Dumas First and foremost, I would like to thank my advisor, Dr.