New Approaches to Aromatic Fluorinations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Docteur» at the University François Rabela
UNIVERSITÉ FRANÇOIS – RABELAIS DE TOURS École Doctorale « Santé - Sciences Biologiques - Chimie du Vivant » and UNIVERSITY OF LJUBLJANA, FACULTY OF PHARMACY «Department of Pharmaceutical Chemistry» A cotutelle thesis submitted in fulfillment of the requirements for the degree of «Docteur» at the University François Rabelais of Tours (France) and Doctor of Pharmacy at the University of Ljubljana (Slovenia) In Pharmaceutical Chemistry Publicly defended on the 1st of March 2013 by Mitja KOVAČ in Ljubljana FLUORATION DE DERIVES DU BENZOVESAMICOL POUR L'OBTENTION DE RADIOLIGANDS POTENTIELS DU TRANSPORTEUR VESICULAIRE DE L'ACETYLCHOLINE Under the co-direction of: Associate Professor Sylvie Mavel (MCU, Tours) and Associate Professor Marko Anderluh (Ljubljana) ----------------- JURY for Oral Defense: Ms MAVEL Sylvie – Associate Professor, University François-Rabelais, Tours, France Mr ANDERLUH Marko – Associate Professor, University of Ljubljana, Slovenia Mr DOLLÉ Frédéric – Service Hospitalier Frédéric Joliot, Institut d'Imagerie BioMédicale - CEA, Orsay, France (Reviewer) Mr EMOND Patrick – Professor, University François-Rabelais, Tours, France Ms GMEINER STOPAR Tanja – Assistant Professor, University of Ljubljana, Slovenia (Reviewer) Mr GOBEC Stanislav – Professor, University of Ljubljana, Slovenia (Chairman) This cotutelle PhD was carried out with the collaboration between the University of Tours (Laboratoire de Biophysique Médicale et Pharmaceutique, Unité INSERM U930 - FRANCE) and the University of Ljubljana (Faculty of Pharmacy, Department of Pharmacutical Chemistry - SLOVENIA). The work was supported by a grant from the Slovene Human Resources Development and Scholarship Fund, by a grant from the University of Ljubljana (Inovativna shema za sofinanciranje doktorskega študija za spodbujanje sodelovanja z gospodarstvom in reševanja aktualnih družbenih izzivov - generacija 2010 Univerza v Ljubljani), and by a Slovenia- French bilateral collaboration project (project n° BI-FR/12-13-PROTEUS-007). -

C-Metalated Nitriles: Diastereoselective Alkylations and Arylations
Duquesne University Duquesne Scholarship Collection Electronic Theses and Dissertations Fall 12-20-2019 C-Metalated Nitriles: Diastereoselective Alkylations and Arylations Robert John Mycka Duquesne University Follow this and additional works at: https://dsc.duq.edu/etd Part of the Organic Chemistry Commons Recommended Citation Mycka, R. J. (2019). C-Metalated Nitriles: Diastereoselective Alkylations and Arylations (Doctoral dissertation, Duquesne University). Retrieved from https://dsc.duq.edu/etd/1851 This Immediate Access is brought to you for free and open access by Duquesne Scholarship Collection. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Duquesne Scholarship Collection. C-METALATED NITRILES: DIASTEREOSELECTIVE ALKYLATIONS AND ARYLATIONS A Dissertation Submitted to the Bayer School of Natural and Environmental Sciences Duquesne University In partial fulfillment of the requirements for the degree of Doctor of Philosophy By Robert J. Mycka December 2019 Copyright by Robert J. Mycka 2019 C-METALATED NITRILES: DIASTEREOSELECTIVE ALKYLATIONS AND ARYLATIONS By Robert J. Mycka Approved November 13, 2019 ________________________________ ________________________________ Dr. Bruce D. Beaver Dr. Jeffrey D. Evanseck Professor of Chemistry and Biochemistry Professor of Chemistry and Biochemistry (Committee Chair) (Committee Member) ________________________________ ________________________________ Dr. Shahed U. M. Khan Dr. Patrick T. Flaherty Associate Professor of Chemistry and -

Friction of Iron Lubricated with Aliphatic and Aromatic Hydrocarbons and Halogenated Analogs
NASA TECHNICAL NOTE 00 0 N w n a c 4 &A 4 a w~.~,z..p~COPY: RETURN TO --;F?.f-..wL TECHNICAL LIBRARY 1(LWTWND ATS, M* M* FRICTION OF IRON LUBRICATED WITH ALIPHATIC AND AROMATIC HYDROCARBONS AND HALOGENATED ANALOGS Donald H. Buckley Lewis Research Center NATIONAL AERONAUTICS AND SPACE ADMINISTRATION WASHINGTON, D. C. APRIL 1976 TECH LIBRARY KAFB,"I .-- - -.- ~~ OL337b7 I 1. Report No. I 2. Government Accession No. ]Recipient's Catalog NO. TN D -8208 I- .- .. I I 4. Title and Subtitle 5. Report Date FRICTION OF IRON LUBRICATED WITH ALIPHATIC AND I April 1976 Performing OrganizationCode AROMATIC HYDROCARBONS AND HALOGENATED ANALOGS I 7. Author(s1 8. Performing Organization Report No. E-8558 Donald H. Buckley ~__ .. 10. Wcrk Unit No. 9. Performing Organization Name and Address 506-16 Lewis Research Center 11. Contract or Grant No. National Aeronautics and Space Administration I Cleveland, Ohio 44135 13. Type of Report and Period Covered ~~ 12. Sponsoring Agency Name and Address Technical Note National Aeronautics and Space Administration 14. Sponsoring Agency Code Washington, D.C. 20546 I 1 15. Supplementary Notes - -- L16. Abstract An investigation was conducted to determine the influence of oxygen and various organic mole cules on the reduction of the friction of an iron (011) single crystal surface. A comparison was made between aliphatic and aromatic structures, all of which contained six carbon atoms, and among various halogen atoms. Results of the investigation indicate that hexane and benzene give similar friction coefficients over a range of loads except at very light loads. At light loads, the friction decreased with an increase in the load where the halogens fluorine and chlorine are in corporated into the benzene molecular structure; however, over the same load range when bro mine and iodine were present, the friction was relatively unchanged. -

Strategies for the Synthesis and Use of Β-Stereogenic Α
STRATEGIES FOR THE SYNTHESIS AND USE OF -STEREOGENIC -KETO ESTERS Kimberly Michelle Steward A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry. Chapel Hill 2012 Approved by: Jeffrey S. Johnson Maurice S. Brookhart Erik J. Alexanian Michel R. Gagné Valerie S. Ashby © 2012 Kimberly Michelle Steward ALL RIGHTS RESERVED ii ABSTRACT KIMBERLY MICHELLE STEWARD: Strategies for the Synthesis and Use of -Stereogenic -Keto Esters (Under the direction of Jeffrey S. Johnson) I. Extant Methods for the Preparation of -Keto Esters An overview of the synthetic methods to prepare -keto esters is presented, with a particular focus on strategies to incorporate a stereocenter at the -position. II. Catalytic Nucleophilic Glyoxylation of Aldehydes The synthesis of -silyloxy -keto esters via a cyanide catalyzed benzoin-type reaction with silyl glyoxylates and aldehydes is described. Critical to the success of the i reaction was identifying Yb(O Pr)3 and acetone cyanohydrin as a mild source of cyanide to prevent isomerization of the products to the corresponding -silyloxy -keto esters. Several secondary transformations add to the utility to the -keto ester products. Of the methods available to prepare -hydroxy -keto acid derivatives, this method provides the most direct route and displays the broadest substrate scope. iii III. Asymmetric Synthesis of -Keto Esters via Cu(II)-Catalyzed Aerobic Deacylation of Acetoacetate Alkylation Products A simple and efficient method for the preparation of -stereogenic -keto esters is described using a copper(II)-catalyzed aerobic deacylation of substituted acetoacetate esters. -

Branched 2-Amino-1,3-Dicyanocyclopenta-1,3-Diene Francisco Ros* Department of Medical Chemistry, Institute of Medical Chemistry, Madrid, Spain
ccines & Va V ACCESS Freely available online f a OPEN o c l c a in n a r t u i o o n J ISSN: 2157-7560 Journal of Vaccines & Vaccination Research Article Branched 2-Amino-1,3-Dicyanocyclopenta-1,3-Diene Francisco Ros* Department of Medical Chemistry, Institute of Medical Chemistry, Madrid, Spain ABSTRACT Reaction of 2-chloroisobutyrophenone with two equivalents of malononitrile anion furnishes 2-amino-1,3- dicyano-5,5-dimethyl-4-phenylcyclopenta-1,3-diene. The cyclic compound represents the novel 2-amino-1,3- dicyanocyclopentadiene structure. The unique 1-cyano-2-amino-3-cyano arrangement in the cyclopentadiene brings about a strong polarization of the electronic configuration of the diene system that is conformed by two opposite dipolar halves. The polarized electronic configuration accounts for the extreme persistence manifested by the cyclopentadiene. The compound owns a vivid lemon-hued yellow color consequent to an unusually intense n absorption of a cyano group in the extensively conjugated compound. This is built up by consecutive one-pot reaction of two molecules of malonon itrile carbanion and the ketonic substrate followed by a new tandem carbon- carbon cyclization with final elimination of cyanate ion. Keywords: 2-Halo ketones; Crowded substitution; Tandem nitrile reaction; Cyclizations; UV/visible spectroscopy; Reaction mechanisms INTRODUCTION this reactant in the course of the reaction, so protecting the reaction yield. Such neutralization could occur by proton transfer from We have been interested in the synthesis of branched-chain organic the emerging alkylated malononitrile having one acidic hydrogen compounds by nucleophilic substitution on activated tertiary easily removable (as contiguous to two cyano groups) to reactant alkyl halides with resonance stabilized carbanions [1-5]. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -
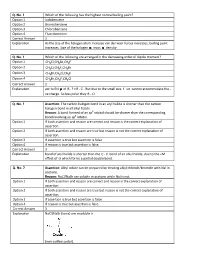
Q. No. 3 Which of the Following Has the Highest Normal Boiling Point
Q. No. 3 Which of the following has the highest normal boiling point? Option 1 Iodobenzene Option 2 Bromobenzene Option 3 Chlorobenzene Option 4 Fluorobenzene Correct Answer 1 Explanation As the size of the halogen atom increase van der waal forces increases, boiling point increases. Size of the halogen mass density. Q. No. 4 Which of the following are arranged in the decreasing order of dipole moment? Option 1 CH3 Cl,CH 3 Br,CH 3 F Option 2 CH3 Cl,CH 3 F,CH 3 Br Option 3 CH3 Br,CH 3 Cl,CH 3 F Option 4 CH3 Br,CH 3 F,CH 3 Cl Correct Answer 2 Explanation acc to EN of RF> RCl . But due to the small size. F ion cannot accommodate the ve charge. So less polar they R Cl . Q. No. 5 Assertion : The carbon halogen bond in an aryl halid e is shorter than the carbon halogen bond in all alkyl halide. Reason : A bond formed of an sp 3 orbital should be shorter than the corresponding bond involving an sp 2 orbital. Option 1 If both assertion and reason are correct and reason is the correct explanation of assertion. Option 2 If both assertion and reason are true but reason is not the correct explanation of assertion. Option 3 If assertion is true but assertion is false. Option 4 If reason is true but assertion is false. Correct Answer 3 Explanation bond of aryl halide is shorter than the C X bond of an alkyl halide, due to the +M effect of X which forms a partial double bond. -

Acenaphthene 83-32-9 5000 N 45000 N 100000 L 110 N 530 N Acephate 30560-19-1 110 N 980 N 2100 N 0.11 N 24 N Acetaldehyde 75-07-0
Table A-6: 2021 Screening Levels Soil Exposure Ground Water Vapor Exposure Chemical Direct Contact Soil MTG Tap Ground Water Indoor Air Residential Com/Ind Excavation Residential Residential Residential Com/Industrial Residential Com/Ind Name CASRN (mg/kg) (mg/kg) (mg/kg) (mg/kg) (ug/L) (ug/L) (ug/L) (ug/m3) (ug/m3) Acenaphthene 83-32-9 5000 N 45000 N 100000 L 110 N 530 N Acephate 30560-19-1 110 N 980 N 2100 N 0.11 N 24 N Acetaldehyde 75-07-0 110 N 340 N 1900 N 0.077 N 19 N 9.4 N 39 N Acetochlor 34256-82-1 1800 N 16000 N 34000 N 5.6 N 350 N Acetone 67-64-1 85000 N 100000 L 100000 L 57 N 14000 N 32000 N 140000 N Acetone Cyanohydrin 75-86-5 100000 L 100000 L 100000 L 2.1 N 8.8 N Acetonitrile 75-05-8 1100 N 3400 N 19000 N 0.54 N 130 N 63 N 260 N Acetophenone 98-86-2 2500 S 2500 S 2500 S 12 N 1900 N Acetylaminofluorene, 2- 53-96-3 2 C 6 C 320 C 0.015 C 0.16 C 0.022 C 0.094 C Acrolein 107-02-8 0.2 N 0.6 N 3.4 N 0.00017 N 0.042 N 0.021 N 0.088 N Acrylamide 79-06-1 3.4 C 46 C 2400 C 0.0021 C 0.5 C 0.1 C 1.2 C Acrylic Acid 79-10-7 140 N 420 N 2300 N 0.0085 N 2.1 N 1 N 4.4 N Acrylonitrile 107-13-1 3.5 C 11 C 370 N 0.0023 C 0.52 C 0.41 C 1.8 C Adiponitrile 111-69-3 100000 L 100000 L 100000 L 6.3 N 26 N Alachlor 15972-60-8 140 C 410 C 18000 N 0.033 M 2 M Aldicarb 116-06-3 88 N 820 N 1800 N 0.015 M 3 M Aldicarb Sulfone 1646-88-4 88 N 820 N 1800 N 0.0088 M 2 M Aldicarb sulfoxide 1646-87-3 0.018 M 4 M Aldrin 309-00-2 0.55 C 1.8 C 59 N 0.03 C 0.0092 C 0.0057 C 0.025 C Allyl Alcohol 107-18-6 4.9 N 15 N 83 N 0.00086 N 0.21 N 0.1 N 0.44 N Allyl Chloride -

Epoxysilane Rearrangement: Mechanistic Studies and Synthetic Applications
Epoxysilane Rearrangement: Mechanistic Studies and Synthetic Applications Michiko Sasaki and Kei Takeda* Department of Synthetic Organic Chemistry, Graduate Schoolof Medical Sciences,Hiroshima University, Received June 28, 2006 Abstract : Mechanistic studies on and synthetic application of epoxysilane rearrangement, a novel synthetic use of ƒ¿, ƒÀ-epoxysilanes, in which an anion-induced ring-opening of epoxide and Brook rearrangement in the resulting ƒ¿-silyl alkoxide occur in a tandem fashion to provide a ƒÀ-siloxy allyl anion, are described. Scheme 2. Attempted asymmetric Brook-rearrangement medi- 1 . Introduction: ated [3 + 2] annulation. Since the first synthesis of ƒ¿, ƒÀ-epoxysilanes reported by Eischl more than forty years ago, many kinds of a, β- epoxysilanes have been synthesized and synthetic method- ology based on their use has been developed.2 The seminal work of Stork and co-workers,3 who showed that a, β-epoxysilanes can be converted to aldehydes and ketones via a ring-opening followed by desilylation, has stimulated considerable interest in their chemistry. In this review we report a mechanistic aspect and synthet- ic applications of epoxysilane rearrangement, which is a novel synthetic use of ƒ¿, ƒÀepoxysilanes and features trans- formation of ƒÀ-silyl-ƒ¿, ƒÀ-epoxy carbanion to J3-siloxy allyl anion. Thirteen years ago, we reported a new approach to the synthesis of highly functionalized cyclopentenol 5 using [3 + 2] annulation that involves a combination of (ƒÀ-(trimethylsi- ment (8 9)(Scheme 2). lyl)acryloyl)silane 1 as the three-carbon unit and lithium This result indicated the possibility of the reaction of an enolate 2 of alkyl methyl ketone as the two-carbon unit, epoxysilane 13 bearing an anion-stabilizing electron-with- which relies on the 1,3-sigmatropic shift in vinylcyclo- drawing group at the a-position with an amide base in the propanolate 4 formed via a 1,2-anionic rearrangement of the presence of an electrophile. -

Ll||L||||||||L|||||||||||||||||||||L||||||L||||||||||||||||||||||||||||||||| US 20030176707A1 (19) United States (12) Patent Application Publication (10) Pub
l|||||||||||||ll||l||||||||l|||||||||||||||||||||l||||||l||||||||||||||||||||||||||||||||| US 20030176707A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0176707 A1 Pye (43) Pub. Date: Sep. 18, 2003 (54) PROCESS FOR PREPARING INTEGRIN Related US. Application Data ANTAGONIST INTERMEDIATE (60) Provisional application No. 60/352,601, ?led on Jan. 29, 2002. (76) Inventor: Philip J. Pye, Guttenberg, NJ (US) Publication Classi?cation Correspondence Address; (51) Int. Cl.7 .................... .. C071) 213/78; C071) 213/72 MERCK AND CO INC (52) US. Cl. .......................................... .. 546/329; 546/330 P O BOX 2000 RAHWAY, NJ 070650907 (57) ABSTRACT (21) Appl, No; 10/353,612 A novel process is provided for the preparation of 2,5-di (3‘-arninopropyl)pyridine Which is useful in the synthesis of otv[33 integrin receptor antagonists. Also provided are useful (22) Filed: Jan. 29, 2003 intermediates obtained from the process. US 2003/0176707 A1 Sep. 18, 2003 PROCESS FOR PREPARING INTEGRIN [0007] The novel process and novel intermediates are ANTAGONIST INTERMEDIATE illustrated in the folloWing embodiment denoted in Scheme 1 beloW. FIELD OF THE INVENTION [0001] The present invention discloses a novel process and Schemel novel intermediates toward the preparation of 2,5-di-(3‘ RZOZC N aminopropyl)pyridine Which is useful in the synthesis of \ MeCN otv[33 integrin receptor antagonists. l base / cozR1 BACKGROUND OF THE INVENTION OM NC N [0002] The present invention provides a novel process for / \ the preparation of 2,5-di-(3‘-aminopropyl)pyridine of struc I Y—X tural formula I. / / CN —> OM (I) OY HZN \ NC N NHZ / / CN _2>H OY [0003] Another aspect of the present invention is con cerned With novel intermediates useful in the disclosed process. -

Chemistry of Allyl Nitrate Esters, Β-Nitroacetamides, and Various Other
Chemistry of Allyl Nitrate Esters, β-Nitroacetamides, and Various Other Nitro Compounds A Thesis Submitted to the Faculty Of Drexel University By Nicholas Paparoidamis in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2013 © Copyright 2013 Nicholas Paparoidamis. All Rights Reserved. ii Acknowledgements I would like to thank Professor Peter Wade for his mentorship and all the other professors and staff who supported me: Dr. Robert Hutchins, Dr. Anthony Addison, Dr. Yen Wei, Dr. Teck-Kah Lim, Dr. David Ruth, Mr. Steve Leesman, Mr. Tim Wade, Mr. Edward Doherty, and so many others. Thank you to every one of my colleagues at Drexel University for their comradeship and support, especially Dr. April Holcomb, Dr. Jonathan Haulenbeek, LT Dr. Christopher Castillo, Arben Kojtari, Noah Johnson, Joshua Smith, and Panagiota Tsetsakos. I would like to most importantly thank my family, Tom Paparoidamis, Fotini Paparoidamis, and Georgia Paparoidamis for all their support during my studies. iii Table of Contents LIST OF TABLES v LIST OF FIGURES vi ABSTRACT vii CHAPTER 1: Tandem Nitration / Rearrangement of Allylic Alcohols 1.1 Introduction 1 1.2 Results and Discussion 22 1.3 Structure Assignments 29 1.4 Experimental 33 1.5 Conclusions 43 CHAPTER 2: Synthesis and Reactions of β-Nitroacetamides 2.1 Introduction 45 2.2 Results and Discussion 52 2.3 Structure Assignments 77 2.4 Experimental 102 2.5 Conclusions 146 CHAPTER 3: Debromination of 2,4-Dibromo-2,4-dinitropentane 3.1 Introduction 148 3.2 Results and Discussion 153 3.3 Structure Assignments 156 3.4 Experimental 157 iv Table of Contents (continued) 3.5 Conclusions 159 LIST OF REFERENCES 160 APPENDIX A: 1H NMR SPECTRA 164 APPENDIX B: 13C NMR SPECTRA 200 VITA 236 v List of Tables 1. -

Process for Preparing Fluorobenzene
European Patent Office 0 Publication number: 0 054 274 Office europeen des brevets A1 tUnUPhAN PATENT APPLICATION (21J Application number: 81110361.3 © Int. CI.3: C 07 C 25/13 C 07 C 17/28 © Dateoffiling: 11.12.81 \m) Priority: 13.1Z.80 JP 176132/80 72) Inventor: Shimokawa, Kazuhiro 28.12.80 JP 186638/80 48-2, Suito-cho Suita-shi Osaka(JP) (43) Date of publication of application: 72) Inventor: Naito, Daiji 23.06.82 Bulletin 82/25 21-6, Takada-cho Ibaraki-shi Osaka(JP) @ Designated Contracting States: DE FR GB §) Inventor: Misaki, Susumu 240-141, Oaza-Aomadani Mino-shi @ Applicant: Daikin Kogyo Co., Ltd. Osaka(JP) No 1-12-39, Umeda Kita-ku Osaka-shi Osaka-fu(JP) Inventor: Yoshida, Tsutomu 2-21-21, Hitotsuya Settsu-shi @ Inventor: Tabushi, Iwao Osaka(JP) 24, Higashisakuragi-cho Matsugasaki Sakyo-ku Kyoto Kyoto(JP) 75) Representative: von Kreisler, Alek, Dipl.-Chem. et al, Deichmannhaus am Hauptbahnhof D-5000 Kolnl(DE) 54) Process for preparing fluorobenzene. Monofluorobenzene is prepared in a good yield by reacting cyclopentadiene or its dimer of the formula: with fluorohalomethane of the formula: wherein Xs are, same or different, halogen atoms in the presence of a phase transfer catalyst and an acid acceptor. This invention relates to a process for preparing fluorobenzene. More particularly, it relates to a process for preparing fluorobenzene by reacting cyclopentadiene or its dimer, bicyclopentadiene, with fluorohalomethane. Fluorobenzene is a very useful compound as a starting material in preparation of medicines, agricultural chemicals,