SPARC Report on the Mystery of Carbon Tetrachloride
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Toxicological Profile for Carbon Tetrachloride
CARBON TETRACHLORIDE 1 1. PUBLIC HEALTH STATEMENT This public health statement tells you about carbon tetrachloride and the effects of exposure to it. The Environmental Protection Agency (EPA) identifies the most serious hazardous waste sites in the nation. These sites are then placed on the National Priorities List (NPL) and are targeted for long-term federal clean-up activities. Carbon tetrachloride has been found in at least 430 of the 1,662 current or former NPL sites. Although the total number of NPL sites evaluated for this substance is not known, the possibility exists that the number of sites at which carbon tetrachloride is found may increase in the future as more sites are evaluated. This information is important because these sites may be sources of exposure and exposure to this substance may harm you. When a substance is released either from a large area, such as an industrial plant, or from a container, such as a drum or bottle, it enters the environment. Such a release does not always lead to exposure. You can be exposed to a substance only when you come in contact with it. You may be exposed by breathing, eating, or drinking the substance, or by skin contact. If you are exposed to carbon tetrachloride, many factors will determine whether you will be harmed. These factors include the dose (how much), the duration (how long), and how you come in contact with it. You must also consider any other chemicals you are exposed to and your age, sex, diet, family traits, lifestyle, and state of health. -
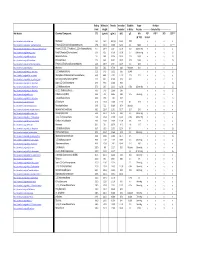
Detectable Compounds by MIP.Pdf
Boiling Molecular Density Ionization Solubility Vapor Analytes Point Weight Potential in Water Pressure ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐Detected By‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Wiki Website Chemical Compound: (oC) (g/mol) (g/mL) (eV) g/L kPa PID* FID** XSD ECD*** @ ~25C 10.6 eV https://en.wikipedia.org/wiki/Methane . Methane ‐161 16.0 .657 g/L 12.61 0.02 n y n n https://en.wikipedia.org/wiki/Dichlorodifluoromethane . Freon 12 (Dichlorodifluoromethane) ‐29.8 120.9 1.486 12.31 0.3 568.0 n y y LS**** https://en.wikipedia.org/wiki/1,1,2‐Trichloro‐1,2,2‐trifluoroethane . Freon 113 (1,1,2‐Trichloro‐1,2,2‐trifluoroethane) 47.7 187.4 1.564 11.78 0.17 285mm Hg n y y y https://en.wikipedia.org/wiki/Vinyl_chloride . Vinyl Chloride (Chloroethene) ‐13.0 62.5 0.910 10.00 2.7 2580mm Hg y y y LS https://en.wikipedia.org/wiki/Bromomethane . Bromomethane 3.6 94.9 3.974 10.53 17.5 190.0 n y y LS https://en.wikipedia.org/wiki/Chloroethane .. Chloroethane 12.3 64.5 0.901 10.97 5.74 134.6 n y y LS https://en.wikipedia.org/wiki/Trichlorofluoromethane . Freon 11 (Trichlorofluoromethane) 23.8 137.4 1.494 11.77 1.1 89.0 n y y y https://en.wikipedia.org/wiki/Acetone . Acetone 56.5 58.1 0.790 9.69 Miscible 30.6 y y n n https://en.wikipedia.org/wiki/1,1‐Dichloroethene . 1,1‐Dichloroethene 32.0 97.0 1.213 9.65 0.04% y y y LS https://en.wikipedia.org/wiki/Dichloromethane . -

Dichlorodifluoromethane Dcf
DICHLORODIFLUOROMETHANE DCF CAUTIONARY RESPONSE INFORMATION 4. FIRE HAZARDS 7. SHIPPING INFORMATION 4.1 Flash Point: 7.1 Grades of Purity: 99.5% (vol.) Common Synonyms Gas Colorless Faint odor Not flammable 7.2 Storage Temperature: Ambient Arcton 6 4.2 Flammable Limits in Air: Not flammable Eskimon 12 7.3 Inert Atmosphere: No requirement 4.3 Fire Extinguishing Agents: Not F-12 Visible vapor cloud is produced. 7.4 Venting: Safety relief flammable Freon 12 7.5 IMO Pollution Category: Currently not available Frigen 12 4.4 Fire Extinguishing Agents Not to Be Genetron 12 Used: Not flammable 7.6 Ship Type: Currently not available Halon 122 4.5 Special Hazards of Combustion 7.7 Barge Hull Type: 3 Isotron 12 Products: Although nonflammable, Ucon 12 dissociation products generated in a fire may be irritating or toxic. 8. HAZARD CLASSIFICATIONS Notify local health and pollution control agencies. 4.6 Behavior in Fire: Helps extinguish fire. 8.1 49 CFR Category: Nonflammable gas Avoid inhalation. 4.7 Auto Ignition Temperature: Not 8.2 49 CFR Class: 2.2 flammable 8.3 49 CFR Package Group: Not pertinent. Not flammable. 4.8 Electrical Hazards: Not pertinent Fire Cool exposed containers with water. 8.4 Marine Pollutant: No 4.9 Burning Rate: Not flammable 8.5 NFPA Hazard Classification: Not listed 4.10 Adiabatic Flame Temperature: Currently 8.6 EPA Reportable Quantity: 5000 pounds CALL FOR MEDICAL AID. not available Exposure 8.7 EPA Pollution Category: D 4.11 Stoichometric Air to Fuel Ratio: Not VAPOR 8.8 RCRA Waste Number: U075 Not irritating to eyes, nose or throat. -

Exposure Investigation Protocol: the Identification of Air Contaminants Around the Continental Aluminum Plant in New Hudson, Michigan Conducted by ATSDR and MDCH
Exposure Investigation Protocol - Continental Aluminum New Hudson, Lyon Township, Oakland County, Michigan Exposure Investigation Protocol: The Identification of Air Contaminants Around the Continental Aluminum Plant in New Hudson, Michigan Conducted by ATSDR and MDCH MDCH/ATSDR - 2004 Exposure Investigation Protocol - Continental Aluminum New Hudson, Lyon Township, Oakland County, Michigan TABLE OF CONTENTS OBJECTIVE/PURPOSE..................................................................................................... 3 RATIONALE...................................................................................................................... 4 BACKGROUND ................................................................................................................ 5 AGENCY ROLES .............................................................................................................. 6 ESTABLISHING CRITERIA ............................................................................................ 7 “Odor Events”................................................................................................................. 7 Comparison Values......................................................................................................... 7 METHODS ....................................................................................................................... 12 Instantaneous (“Grab”) Air Sampling........................................................................... 12 Continuous Air Monitoring.......................................................................................... -

Refrigerant Safety Refrigerant History
Refrigerant Safety The risks associated with the use of refrigerants in refrigeration and airconditioning equipment can include toxicity, flammability, asphyxiation, and physical hazards. Although refrigerants can pose one or more of these risks, system design, engineering controls, and other techniques mitigate this risk for the use of refrigerant in various types of equipment. Refrigerant History Nearly all of the historically used refrigerants were flammable, toxic, or both. Some were also highly reactive, resulting in accidents (e. g., leak, explosion) due to equipment failure, poor maintenance, or human error. The task of finding a nonflammable refrigerant with good stability was given to Thomas Midgley in 1926. With his associates Albert Leon Henne and Robert Reed McNary, Dr. Midgley observed that the refrigerants then in use comprised relatively few chemical elements, many of which were clustered in an intersecting row and column of the periodic table of elements. The element at the intersection was fluorine, known to be toxic by itself. Midgley and his collaborators felt, however, that compounds containing fluorine could be both nontoxic and nonflammable. The attention of Midgley and his associates was drawn to organic fluorides by an error in the literature that showed the boiling point for tetrafluoromethane (carbon tetrafluoride) to be high compared to those for other fluorinated compounds. The correct boiling temperature later was found to be much lower. Nevertheless, the incorrect value was in the range sought and led to evaluation of organic fluorides as candidates. The shorthand convention, later introduced to simplify identification of the organic fluorides for a systematic search, is used today as the numbering system for refrigerants. -

Summary of Gas Cylinder and Permeation Tube Standard Reference Materials Issued by the National Bureau of Standards
A111D3 TTbS?? o z C/J NBS SPECIAL PUBLICATION 260-108 o ^EAU U.S. DEPARTMENT OF COMMERCE/National Bureau of Standards Standard Reference Materials: Summary of Gas Cylinder and Permeation Tube Standard Reference Materials Issued by the National Bureau of Standards QC 100 U57 R. Mavrodineanu and T. E. Gills 260-108 1987 m he National Bureau of Standards' was established by an act of Congress on March 3, 1901. The Bureau's overall goal i s t0 strengthen and advance the nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research to assure international competitiveness and leadership of U.S. industry, science arid technology. NBS work involves development and transfer of measurements, standards and related science and technology, in support of continually improving U.S. productivity, product quality and reliability, innovation and underlying science and engineering. The Bureau's technical work is performed by the National Measurement Laboratory, the National Engineering Laboratory, the Institute for Computer Sciences and Technology, and the Institute for Materials Science and Engineering. The National Measurement Laboratory Provides the national system of physical and chemical measurement; • Basic Standards 2 coordinates the system with measurement systems of other nations and • Radiation Research furnishes essential services leading to accurate and uniform physical and • Chemical Physics chemical measurement throughout the Nation's scientific community, • Analytical Chemistry industry, and commerce; provides advisory and research services to other Government agencies; conducts physical and chemical research; develops, produces, and distributes Standard Reference Materials; provides calibration services; and manages the National Standard Reference Data System. -

Flammable Refrigerants Firefighter Training
Flammable refrigerants firefighter training: Hazard assessment and demonstrative testing FINAL REPORT BY: Noah L. Ryder, P. E. Stephen J. Jordan Fire & Risk Alliance, LLC. Rockville, Maryland, USA. Peter B. Sunderland, Ph.D. University of Maryland Department of Fire Protection Engineering College Park, Maryland, USA. May 2019 © 2019 Fire Protection Research Foundation 1 Batterymarch Park, Quincy, MA 02169-7417, USA Email: [email protected] | Web: nfpa.org/foundation ---page intentionally left blank--- —— Page ii —— FOREWORD The ongoing push toward sustainability of refrigeration systems will require the adoption of low global warming potential (GWP) refrigerants to meet the shift in environmental regulations. Fire safety is a lingering issue with the new age of flammable refrigerants being adopted and first responders may not be familiar with the change in material hazards or the appropriate response procedures required to safely handle these fire scenarios. This project is part of the overall two-year project with a goal to enhance firefighter safety and reduce potential injury by providing training on the hazards from appliances with flammable refrigerants. It will document the information about flammable refrigerants technologies and the hazards to emergency responders and develop interactive training modules to transfer the knowledge to the fire service. This report is focused to develop material documenting the hazards associated with flammable refrigerant technologies and the risks posed to the first responders. The material documentation included a literature review, Task 1, identifying baseline information on flammable refrigerants, their existing usage and implementation into products, potential integration into future technologies, and finally any existing guidance and best practices on response and tactics. -

Carbon Tetrachloride
CARBON TETRACHLORIDE Data were last reviewed in IARC (1979) and the compound was classified in IARC Monographs Supplement 7 (1987a). 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Nomenclature Chem. Abstr. Serv. Reg. No.: 56-23-5 Chem. Abstr. Name: Tetrachloromethane IUPAC Systematic Name: Carbon tetrachloride Synonyms: Benzinoform; carbona 1.1.2 Structural and molecular formulae and relative molecular mass Cl Cl CCl Cl CCl4 Relative molecular mass: 153.82 1.1.3 Chemical and physical properties of the pure substance (a) Description: Colourless, clear, nonflammable, liquid with a characteristic odour (Budavari, 1996) (b) Boiling-point: 76.8°C (Lide, 1997) (c) Melting-point: –23°C (Lide, 1997) (d) Solubility: Very slightly soluble in water (0.05% by volume); miscible with benzene, chloroform, diethyl ether, carbon disulfide and ethanol (Budavari, 1996) (e) Vapour pressure: 12 kPa at 20°C; relative vapour density (air = 1), 5.3 at the boiling-point (American Conference of Governmental Industrial Hygienists, 1991) (f) Conversion factor: mg/m3 = 6.3 × ppm 1.2 Production and use Production in the United States in 1991 was reported to be approximately 143 thousand tonnes (United States International Trade Commission, 1993). Information –401– 402 IARC MONOGRAPHS VOLUME 71 available in 1995 indicated that carbon tetrachloride was produced in 24 countries (Che- mical Information Services, 1995). Carbon tetrachloride is used in the synthesis of chlorinated organic compounds, including chlorofluorocarbon refrigerants. It is also used as an agricultural fumigant and as a solvent in the production of semiconductors, in the processing of fats, oils and rubber and in laboratory applications (Lewis, 1993; Kauppinen et al., 1998). -

Toxicological Profile for Carbon Tetrachloride
CARBON TETRACHLORIDE 171 4. CHEMICAL AND PHYSICAL INFORMATION 4.1 CHEMICAL IDENTITY Information regarding the chemical identity of carbon tetrachloride is located in Table 4-1. 4.2 PHYSICAL AND CHEMICAL PROPERTIES Information regarding the physical and chemical properties of carbon tetrachloride is located in Table 4-2. CARBON TETRACHLORIDE 172 4. CHEMICAL AND PHYSICAL INFORMATION Table 4-1. Chemical Identity of Carbon Tetrachloride Characteristic Information Reference Chemical name Carbon tetrachloride IARC 1979 Synonym(s) Carbona; carbon chloride; carbon tet; methane tetrachloride; HSDB 2004 perchloromethane; tetrachloromethane; benzinoform Registered trade name(s) Benzinoform; Fasciolin; Flukoids; Freon 10; Halon 104; IARC 1979 Tetraform; Tetrasol Chemical formula CCl4 IARC 1979 Chemical structure Cl IARC 1979 Cl Cl Cl Identification numbers: CAS registry 56-23-5 NLM 1988 NIOSH RTECS FG4900000 HSDB 2004 EPA hazardous waste U211; D019 HSDB 2004 OHM/TADS 7216634 HSDB 2004 DOT/UN/NA/IMCO UN1846; IMCO 6.1 HSDB 2004 shipping HSDB 53 HSDB 2004 NCI No data CAS = Chemical Abstracts Service; DOT/UN/NA/IMCO = Department of Transportation/United Nations/North America/International Maritime Dangerous Goods Code; EPA = Environmental Protection Agency; HSDB = Hazardous Substances Data Bank; NCI = National Cancer Institute; NIOSH = National Institute for Occupational Safety and Health; OHM/TADS = Oil and Hazardous Materials/Technical Assistance Data System; RTECS = Registry of Toxic Effects of Chemical Substances CARBON TETRACHLORIDE 173 4. CHEMICAL -
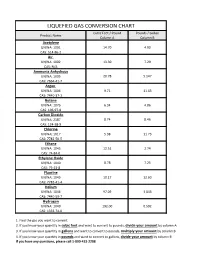
Liquefied Gas Conversion Chart
LIQUEFIED GAS CONVERSION CHART Cubic Feet / Pound Pounds / Gallon Product Name Column A Column B Acetylene UN/NA: 1001 14.70 4.90 CAS: 514-86-2 Air UN/NA: 1002 13.30 7.29 CAS: N/A Ammonia Anhydrous UN/NA: 1005 20.78 5.147 CAS: 7664-41-7 Argon UN/NA: 1006 9.71 11.63 CAS: 7440-37-1 Butane UN/NA: 1075 6.34 4.86 CAS: 106-97-8 Carbon Dioxide UN/NA: 2187 8.74 8.46 CAS: 124-38-9 Chlorine UN/NA: 1017 5.38 11.73 CAS: 7782-50-5 Ethane UN/NA: 1045 12.51 2.74 CAS: 74-84-0 Ethylene Oxide UN/NA: 1040 8.78 7.25 CAS: 75-21-8 Fluorine UN/NA: 1045 10.17 12.60 CAS: 7782-41-4 Helium UN/NA: 1046 97.09 1.043 CAS: 7440-59-7 Hydrogen UN/NA: 1049 192.00 0.592 CAS: 1333-74-0 1. Find the gas you want to convert. 2. If you know your quantity in cubic feet and want to convert to pounds, divide your amount by column A 3. If you know your quantity in gallons and want to convert to pounds, multiply your amount by column B 4. If you know your quantity in pounds and want to convert to gallons, divide your amount by column B If you have any questions, please call 1-800-433-2288 LIQUEFIED GAS CONVERSION CHART Cubic Feet / Pound Pounds / Gallon Product Name Column A Column B Hydrogen Chloride UN/NA: 1050 10.60 8.35 CAS: 7647-01-0 Krypton UN/NA: 1056 4.60 20.15 CAS: 7439-90-9 Methane UN/NA: 1971 23.61 3.55 CAS: 74-82-8 Methyl Bromide UN/NA: 1062 4.03 5.37 CAS: 74-83-9 Neon UN/NA: 1065 19.18 10.07 CAS: 7440-01-9 Mapp Gas UN/NA: 1060 9.20 4.80 CAS: N/A Nitrogen UN/NA: 1066 13.89 6.75 CAS: 7727-37-9 Nitrous Oxide UN/NA: 1070 8.73 6.45 CAS: 10024-97-2 Oxygen UN/NA: 1072 12.05 9.52 CAS: 7782-44-7 Propane UN/NA: 1075 8.45 4.22 CAS: 74-98-6 Sulfur Dioxide UN/NA: 1079 5.94 12.0 CAS: 7446-09-5 Xenon UN/NA: 2036 2.93 25.51 CAS: 7440-63-3 1. -

Product Stewardship Summary Carbon Tetrachloride
Product Stewardship Summary Carbon Tetrachloride May 31, 2017 version Summary This Product Stewardship Summary is intended to give general information about Carbon Tetrachloride. It is not intended to provide an in-depth discussion of all health and safety information about the product or to replace any required regulatory communications. Carbon tetrachloride is a colorless liquid with a sweet smell that can be detected at low levels; however, odor is not a reliable indicator that occupational exposure levels have not been exceeded. Its chemical formula is CC14. Most emissive uses of carbon tetrachloride, a “Class I” ozone-depleting substance (ODS), have been phased out under the Montreal Protocol on Substances that Deplete the Ozone Layer (Montreal Protocol). Certain industrial uses (e.g. feedstock/intermediate, essential laboratory and analytical procedures, and process agent) are allowed under the Montreal Protocol. 1. Chemical Identity Name: carbon tetrachloride Synonyms: tetrachloromethane, methane tetrachloride, perchloromethane, benzinoform Chemical Abstracts Service (CAS) number: 56-23-5 Chemical Formula: CC14 Molecular Weight: 153.8 Carbon tetrachloride is a colorless liquid. It has a sweet odor, and is practically nonflammable at ambient temperatures. 2. Production Carbon tetrachloride can be manufactured by the chlorination of methyl chloride according to the following reaction: CH3C1+ 3C12 —> CC14 + 3HC1 Carbon tetrachloride can also be formed as a byproduct of the manufacture of chloromethanes or perchloroethylene. OxyChem manufactures carbon tetrachloride at facilities in Geismar, Louisiana and Wichita, Kansas. Page 1 of 7 3. Uses The use of carbon tetrachloride in consumer products is banned by the Consumer Product Safety Commission (CPSC), under the Federal Hazardous Substance Act (FHSA) 16 CFR 1500.17. -

13C NMR Study of Co-Contamination of Clays with Carbon Tetrachloride
Environ. Sci. Technol. 1998, 32, 350-357 13 sometimes make it the equal of solid acids like zeolites or C NMR Study of Co-Contamination silica-aluminas. Benesi (7-9) measured the Hammett acidity of Clays with Carbon Tetrachloride function H0 for a number of clays; these H0 values range from +1.5 to -8.2 (in comparison to H0 )-12 for 100% ) and Benzene sulfuric acid and H0 5 for pure acetic acid). Therefore, one can expect that certain chemical transformations might occur in/on clays that are similar to what are observed in zeolite TING TAO AND GARY E. MACIEL* systems. Thus, it is of interest to examine what happens Department of Chemistry, Colorado State University, when carbon tetrachloride and benzene are ªco-contami- Fort Collins, Colorado 80523 nantsº in a clay. This kind of information would be useful in a long-term view for understanding chemical transforma- tions of contaminants in soil at contaminated sites. Data on 13 these phenomena could also be useful for designing predic- Both solid-sample and liquid-sample C NMR experiments tive models and/or effective pollution remediation strategies. have been carried out to identify the species produced by Solid-state NMR results, based on 13C detection and line the reaction between carbon tetrachloride and benzene narrowing by magic angle spinning (MAS) and high-power when adsorbed on clays, kaolinite, and montmorillonite. Liquid- 1H decoupling (10), have been reported on a variety of organic sample 13C and 1H NMR spectra of perdeuteriobenzene soil components such as humic samples (11-13). Appar- extracts confirm the identities determined by solid-sample ently, there have been few NMR studies concerned directly 13C NMR and provide quantitative measures of the amounts with elucidating the interactions of organic compounds with of the products identifiedsbenzoic acid, benzophenone, and soil or its major components.