Front Matter Template
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Open Matthew R Moreau Ph.D. Dissertation Finalfinal.Pdf
The Pennsylvania State University The Graduate School Department of Veterinary and Biomedical Sciences Pathobiology Program PATHOGENOMICS AND SOURCE DYNAMICS OF SALMONELLA ENTERICA SEROVAR ENTERITIDIS A Dissertation in Pathobiology by Matthew Raymond Moreau 2015 Matthew R. Moreau Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2015 The Dissertation of Matthew R. Moreau was reviewed and approved* by the following: Subhashinie Kariyawasam Associate Professor, Veterinary and Biomedical Sciences Dissertation Adviser Co-Chair of Committee Bhushan M. Jayarao Professor, Veterinary and Biomedical Sciences Dissertation Adviser Co-Chair of Committee Mary J. Kennett Professor, Veterinary and Biomedical Sciences Vijay Kumar Assistant Professor, Department of Nutritional Sciences Anthony Schmitt Associate Professor, Veterinary and Biomedical Sciences Head of the Pathobiology Graduate Program *Signatures are on file in the Graduate School iii ABSTRACT Salmonella enterica serovar Enteritidis (SE) is one of the most frequent common causes of morbidity and mortality in humans due to consumption of contaminated eggs and egg products. The association between egg contamination and foodborne outbreaks of SE suggests egg derived SE might be more adept to cause human illness than SE from other sources. Therefore, there is a need to understand the molecular mechanisms underlying the ability of egg- derived SE to colonize the chicken intestinal and reproductive tracts and cause disease in the human host. To this end, the present study was carried out in three objectives. The first objective was to sequence two egg-derived SE isolates belonging to the PFGE type JEGX01.0004 to identify the genes that might be involved in SE colonization and/or pathogenesis. -

Fucosyltransferase 8 As a Functional Regulator of Nonsmall Cell Lung Cancer
Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer Chien-Yu Chena,b, Yi-Hua Janb, Yi-Hsiu Juanc, Chih-Jen Yangd, Ming-Shyan Huangd, Chong-Jen Yuc, Pan-Chyr Yangc, Michael Hsiaob, Tsui-Ling Hsub,1, and Chi-Huey Wongb,1 aInstitute of Biochemical Sciences, National Taiwan University, Taipei 106, Taiwan; bGenomics Research Center, Academia Sinica, Taipei 115, Taiwan; cDepartment of Internal Medicine, National Taiwan University Hospital, Taipei 100, Taiwan; and dDepartment of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 807, Taiwan Contributed by Chi-Huey Wong, November 26, 2012 (sent for review August 17, 2012) The up-regulation of fucosyltransferase 8 (FUT8), the only enzyme downstream signaling. Furthermore, the increase in core fuco- catalyzing α1,6-fucosylation in mammals, has been observed in sylation on E-cadherin has been shown to strengthen cell–cell several malignant cancers including liver, ovarian, thyroid, and co- adhesion (10). lorectal cancers. However, the pathological role and the regulatory Both transgenic and knockout mouse models have been gen- mechanism of FUT8 in cancers remain largely unknown. In the cur- erated to study the physiological role of FUT8 (7, 11, 12). Ec- rent study, we report that the expression of FUT8 is up-regulated in topic expression of FUT8 in mice results in an accumulation of nonsmall cell lung cancer (NSCLC) and correlates with tumor me- lipid droplets in hepatocytes and proximal renal tubular cells. tastasis, disease recurrence, and poor survival in patients with This steatosis-like phenotype observed in transgenic mice is NSCLC. Knocking down FUT8 in aggressive lung cancer cell lines linked to the activity of liver lysosomal acid lipase, which becomes significantly inhibits their malignant behaviors including in vitro inactive when over core-fucosylated (11), suggesting that excess invasion and cell proliferation, as well as in vivo metastasis and core fucosylation may lead to a breakdown of normal lipid me- tumor growth. -

Estudi Cinètic De L'activitat Enzimàtica De GT-MG517
PART EXPERIMENTAL - Materials i mètodes – - 229 - - 230 - - 231 - - 232 - Capítol 8: Protocols de biologia molecular - 233 - - 234 - Capítol 8: Protocols de biologia molecular 8 Protocols de biologia molecular 8.1 Capítol 2: Estudi de l’essencialitat de les possibles glicosiltransferases de Mycoplasma genitalium 8.1.1 Preparació de medi SP4 La preparació de medi SP4, medi complex que s’usa per al creixement de Mycoplasma genitalium, té lloc com s’explica a continuació. A la Taula 8.1 se’n presenten els components i els volums corresponents per a la preparació de 500 mL de medi líquid. Taula 8.1. Composició i preparació del medi SP4. Quantitat per Compost a 500 mL Preparació de la base PPLO (Difco) 1.75 g Triptona (Difco) 5 g Bactopeptona (Difco) 2.65 g Glucosa (Sigma-Aldrich) 2.5 g Aigua destil·lada Fins a 312 mL Preparació dels complements Yestolate 2 % autoclavat (Difco) 50 mL Roig de fenol 0.1 % pH 7 autoclavat 7 mL Extracte de llevat fresc 25 % 17.5 mL CMRL 10x (Invitrogen) 25 mL Sèrum boví fetal (Invitrogen) 85 mL Glutamina 29.2 mg/mL 1.71 mL Inicialment es mesclen tots els components corresponents a la base del medi i s’ajusta el pH a 7.8 amb NaOH 1 M. El conjunt s’autoclava 15 min a 120 ºC i es deixa refredar abans d’afegir-hi els complements. Un cop aquests s’han addicionat, cal comprovar que el pH del medi es manté entre 7.6 i 7.8. La preparació de l’extracte de llevat té lloc a partir de la dilució de 250 g de llevat fresc en 1 L d’aigua destil·lada. -
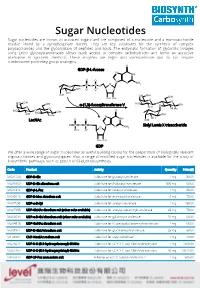
Sugar Nucleotides Sugar Nucleotides Are Known As Activated Sugars and Are Composed of a Nucleoside and a Monosaccharide Residue Linked by a Pyrophosphate Moiety
Sugar Nucleotides Sugar nucleotides are known as activated sugars and are composed of a nucleoside and a monosaccharide residue linked by a pyrophosphate moiety. They are key substrates for the synthesis of complex polysaccharides and the glycosylation of peptides and lipids. The enzymatic formation of glycosidic linkages using Leloir glycosyltransferases allows quick access to complex carbohydrates and forms an attractive alternative to synthetic methods. These enzymes are regio- and stereoselective and do not require cumbersome protecting group strategies. GDP-β-L-fucose α-(1,3)-fucosyltransferase V LacNAc Sialyl Lewis X trisaccharide GDP We offer a wide range of sugar nucleotides as useful building blocks for the preparation of biologically relevant oligosaccharides and glycoconjugates. Also, a range of modified sugar nucleotides is available for the study of biosynthetic pathways, such as Lipid A or (Sia)LeX biosynthesis. Code Product Activity Quantity Price ($) MG31129 GDP-D-Glc Substrate for glucosyltransferase 1 mg 50.00 MU08960 UDP-D-Glc disodium salt Substrate for β-glucosyltransferase 500 mg 60.00 MG01912 GDP-β-L-Fuc Substrate for fucosyltransferase 1 mg 85.00 MG05610 GDP-D-Man disodium salt Substrate for mannosyltransferase 5 mg 75.00 MU07658 UDP-α-D-Xyl Substrate for xylosyltransferase 1 mg 150.00 MU07955 UDP-GlcNAc disodium salt (other salts available) Substrate for acetylglucosaminyltransferase 25 mg 75.00 MU06699 UDP-α-D-Gal disodium salt (other salts available) Substrate for galactosyltransferase 10 mg 68.30 MU04515 UDP-GalNAc -

Advances in Understanding Glycosyltransferases from A
Available online at www.sciencedirect.com ScienceDirect Advances in understanding glycosyltransferases from a structural perspective Tracey M Gloster Glycosyltransferases (GTs), the enzymes that catalyse commonly activated nucleotide sugars, but can also be glycosidic bond formation, create a diverse range of lipid phosphates and unsubstituted phosphate. saccharides and glycoconjugates in nature. Understanding GTs at the molecular level, through structural and kinetic GTs have been classified by sequence homology into studies, is important for gaining insights into their function. In 96 families in the Carbohydrate Active enZyme data- addition, this understanding can help identify those enzymes base (CAZy) [1 ]. The CAZy database provides a highly which are involved in diseases, or that could be engineered to powerful predictive tool, as the structural fold and synthesize biologically or medically relevant molecules. This mechanism of action are invariant in most of the review describes how structural data, obtained in the last 3–4 families. Therefore, where the structure and mechanism years, have contributed to our understanding of the of a GT member for a given family has been reported, mechanisms of action and specificity of GTs. Particular some assumptions about other members of the family highlights include the structure of a bacterial can be made. Substrate specificity, however, is more oligosaccharyltransferase, which provides insights into difficult to predict, and requires experimental charac- N-linked glycosylation, the structure of the human O-GlcNAc terization of individual GTs. Determining both the transferase, and the structure of a bacterial integral membrane sugar donor and acceptor for a GT of unknown function protein complex that catalyses the synthesis of cellulose, the can be challenging, and is one of the reasons there are most abundant organic molecule in the biosphere. -

GDP-Glo™ Glycosyltransferase Assay Instructions for Use of Products VA1090, VA1091 and VA1092
TECHNICAL MANUAL GDP-Glo™ Glycosyltransferase Assay Instructions for Use of Products VA1090, VA1091 and VA1092 10/17 TM505 GDP-Glo™ Glycosyltransferase Assay All technical literature is available at: www.promega.com/protocols/ Visit the web site to verify that you are using the most current version of this Technical Manual. E-mail Promega Technical Services if you have questions on use of this system: [email protected] 1. Description .........................................................................................................................................1 2. Product Components and Storage Conditions ........................................................................................6 3. Preparing for the GDP-Glo™ Glycosyltransferase Assay .........................................................................9 3.A. Preparing the GDP Detection Reagent ..........................................................................................9 3.B. Generating a Standard Curve for GDP ........................................................................................ 10 4. GDP-Glo™ Glycosyltransferase Assay Protocols ................................................................................... 11 4.A. GDP-Glo™ Glycosyltransferase Assay Protocol ............................................................................ 11 4.B. Optimizing Glycosyltransferase Reaction Conditions ................................................................... 12 4.C. Determining Km Value for FUT7 Substrate GDP-Fucose .............................................................. -

Ep 1 117 822 B1
Europäisches Patentamt (19) European Patent Office & Office européen des brevets (11) EP 1 117 822 B1 (12) EUROPÄISCHE PATENTSCHRIFT (45) Veröffentlichungstag und Bekanntmachung des (51) Int Cl.: Hinweises auf die Patenterteilung: C12P 19/18 (2006.01) C12N 9/10 (2006.01) 03.05.2006 Patentblatt 2006/18 C12N 15/54 (2006.01) C08B 30/00 (2006.01) A61K 47/36 (2006.01) (21) Anmeldenummer: 99950660.3 (86) Internationale Anmeldenummer: (22) Anmeldetag: 07.10.1999 PCT/EP1999/007518 (87) Internationale Veröffentlichungsnummer: WO 2000/022155 (20.04.2000 Gazette 2000/16) (54) HERSTELLUNG VON POLYGLUCANEN DURCH AMYLOSUCRASE IN GEGENWART EINER TRANSFERASE PREPARATION OF POLYGLUCANS BY AMYLOSUCRASE IN THE PRESENCE OF A TRANSFERASE PREPARATION DES POLYGLUCANES PAR AMYLOSUCRASE EN PRESENCE D’UNE TRANSFERASE (84) Benannte Vertragsstaaten: (56) Entgegenhaltungen: AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU WO-A-00/14249 WO-A-00/22140 MC NL PT SE WO-A-95/31553 (30) Priorität: 09.10.1998 DE 19846492 • OKADA, GENTARO ET AL: "New studies on amylosucrase, a bacterial.alpha.-D-glucosylase (43) Veröffentlichungstag der Anmeldung: that directly converts sucrose to a glycogen- 25.07.2001 Patentblatt 2001/30 like.alpha.-glucan" J. BIOL. CHEM. (1974), 249(1), 126-35, XP000867741 (73) Patentinhaber: Südzucker AG Mannheim/ • BUTTCHER, VOLKER ET AL: "Cloning and Ochsenfurt characterization of the gene for amylosucrase 68165 Mannheim (DE) from Neisseria polysaccharea: production of a linear.alpha.-1,4-glucan" J. BACTERIOL. (1997), (72) Erfinder: 179(10), 3324-3330, XP002129879 • GALLERT, Karl-Christian • DE MONTALK, G. POTOCKI ET AL: "Sequence D-61184 Karben (DE) analysis of the gene encoding amylosucrase • BENGS, Holger from Neisseria polysaccharea and D-60598 Frankfurt am Main (DE) characterization of the recombinant enzyme" J. -

Analyzing the Complex Machinery of Cell Wall Biosynthesis
Analyzing the complex machinery of cell wall biosynthesis Jaap Timmers Thesis committee Thesis supervisor Prof. dr. R.G.F. Visser Professor of Plant Breeding Wageningen University Thesis co-supervisor Dr.ir. L.M. Trindade Assistant professor at the Laboratory of Plant Breeding Wageningen University Dr.ir. J.-P. Vincken Assistant professor at the Laboratory of Food Chemistry Wageningen University Other members Prof.dr.ir. A.G.J. Voragen Wageningen University Dr. H. Höfte INRA Centre de Versailles, France Dr.ir. G.H. Immink Plant Research International Prof.dr. M.E. Janson Wageningen University This research was conducted under the auspices of the Graduate School of Experimental Plant Sciences. Analyzing the complex machinery of cell wall biosynthesis Jaap Timmers Thesis submitted in partial fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof.dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Doctorate Board to be defended in public on Friday 4 December 2009 at 11 AM in the Aula. Timmers, J.F.P Analyzing the complex machinery of cell wall biosynthesis Ph.D. thesis Wageningen University, the Netherlands, 2009 with summary in Dutch ISBN 978-90-8585-516-3 Contents Chapter 1 7 General introduction Chapter 2 21 Implementation and optimization of a system to characterize protein complexes involved in plant cell wall biosynthesis Chapter 3 35 Interactions between membrane bound cellulose synthases involved in the synthesis of the secondary cell -

Synthesis of Human Milk Oligosaccharides: Protein Engineering Strategies for Improved Enzymatic Transglycosylation
molecules Review Synthesis of Human Milk Oligosaccharides: Protein Engineering Strategies for Improved Enzymatic Transglycosylation Birgitte Zeuner , David Teze , Jan Muschiol and Anne S. Meyer * Protein Chemistry and Enzyme Technology, Department of Biotechnology and Biomedicine, Technical University of Denmark, 2800 Kgs Lyngby, Denmark; [email protected] (B.Z.); [email protected] (D.T.); [email protected] (J.M.) * Correspondence: [email protected]; Tel.: +45-45252600 Academic Editor: Ramón J. Estévez Cabanas Received: 30 April 2019; Accepted: 26 May 2019; Published: 28 May 2019 Abstract: Human milk oligosaccharides (HMOs) signify a unique group of oligosaccharides in breast milk, which is of major importance for infant health and development. The functional benefits of HMOs create an enormous impetus for biosynthetic production of HMOs for use as additives in infant formula and other products. HMO molecules can be synthesized chemically, via fermentation, and by enzymatic synthesis. This treatise discusses these different techniques, with particular focus on harnessing enzymes for controlled enzymatic synthesis of HMO molecules. In order to foster precise and high-yield enzymatic synthesis, several novel protein engineering approaches have been reported, mainly concerning changing glycoside hydrolases to catalyze relevant transglycosylations. The protein engineering strategies for these enzymes range from rationally modifying specific catalytic residues, over targeted subsite 1 mutations, to unique and novel transplantations of designed − peptide sequences near the active site, so-called loop engineering. These strategies have proven useful to foster enhanced transglycosylation to promote different types of HMO synthesis reactions. The rationale of subsite 1 modification, acceptor binding site matching, and loop engineering, − including changes that may alter the spatial arrangement of water in the enzyme active site region, may prove useful for novel enzyme-catalyzed carbohydrate design in general. -

Function Study of Arabidopsis Thaliana Core Alpha1,3-Fucosyltransferase (Fucta) Peter Both
Structure – function study of Arabidopsis thaliana core alpha1,3-fucosyltransferase (FucTA) Peter Both To cite this version: Peter Both. Structure – function study of Arabidopsis thaliana core alpha1,3-fucosyltransferase (FucTA). Biomolecules [q-bio.BM]. Université Joseph-Fourier - Grenoble I, 2009. English. tel- 00449431 HAL Id: tel-00449431 https://tel.archives-ouvertes.fr/tel-00449431 Submitted on 21 Jan 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITE JOSEPH FOURIER (GRENOBLE I ) ECOLE DOCTORALE CHIMIE ET SCIENCES DU VIVANT UNIVERSITE SLOVAQUE DE TECHNOLOGIE DE BRATISLAVA (STUBA) THESE Pour l’obtention du Diplôme de DOCTEUR DE L’UNIVERSITE JOSEPH FOURIER Discipline: Biologie Présentée et soutenue publiquement le 29 octobre 2009 par PETER BOTH ETUDE STRUCTURE-FUNCTION D’UNE FUCOSYLTRANSFERASE (FUCT-A) DE ARABIDOPSIS THALIANA JURY Rapporteurs : Prof. Abderrahman MAFTAH, Université de Limoges Dr Eva HOSTINOVA, Slovak Academy of Sciences, Bratislava Examinateurs : Dr Eva KUTEJOVA, Slovak Academy of Sciences, Bratislava Dr Serge PEREZ, ESRF, Grenoble Dr Jan MUCHA, Slovak Academy of Sciences, Bratislava Prof. Christelle BRETON, Université Grenoble I Thèse préparée au CERMAV (Grenoble) et ICHSAS (Bratislava) 1 I would like to express my gratitude to all those who gave me the possibility to complete this thesis. -

A Cloned Human Cdna Determines Expression of a Mouse Stage-Specific Embryonic Antigen and the Lewis Blood Group (1,3/1,4)-Fucosyltransferase
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press A cloned human cDNA determines expression of a mouse stage-specific embryonic antigen and the Lewis blood group (1,3/1,4)-fucosyltransferase Jolanta F. Kukowska-Latallo, ~ Robert D. Larsen, ~ Rajan P. Nair, ~ and John B. Lowe ~,2,s tHoward Hughes Medical Institute and the Department of Pathology, The University of Michigan Medical School, Ann Arbor, Michigan 48109-0650 USA The stage-specific embryonic antigen SSEA-I is a cell-surface oligosaccharide molecule expressed with temporal precision during the murine preimplantation period and implicated in adhesive events involving the process of compaction. We used a mammalian transient expression system to isolate a cloned human cDNA that determines expression of the SSEA-I molecule. The cDNA sequence predicts a type II transmembrane protein with a domain structure similar to mammalian glycosyltransferases, but without primary sequence similarity to these enzymes. The carboxy-terminal domain of this protein was shown to be catalytically active as a fucosyltransferase when expressed in COS-I cells as a portion of a secreted protein A fusion peptide. The enzyme is an exceptional glycosyltransferase in that it can use both type I and type II oligosaccharides as acceptor substrates to generate subterminal Fucc~(l,4)- and Fuc~(l,3)-linkages, respectively, in a manner analogous to the human Lewis blood group fucosyltransferase. Southern blot analysis shows that the cDNA corresponds to sequences syntenic to the Lewis locus on chromosome 19. These results indicate that this cDNA is the product of the human Lewis blood group locus, provide genetic confirmation of the hypothesis that this enzyme can catalyze two distinct transglycosylation reactions, and outline an approach to the isolation of other sequences that determine expression of developmentally regulated oligosaccharide antigens. -

Biochemical Characteristics and Function of a Fucosyltransferase Encoded by Ste7 in Ebosin Biosynthesis of Streptomyces Sp. 139
J. Microbiol. Biotechnol. (2009), 19(10), 1092–1097 doi: 10.4014/jmb.0903.03021 First published online 8 June 2009 Biochemical Characteristics and Function of a Fucosyltransferase Encoded by ste7 in Ebosin Biosynthesis of Streptomyces sp. 139 1 1 2 1 1 1 3 Chang, Ming , Li-Ping Bai , Jung-Jie Shan , Rong Jiang , Yang Zhang , Lian-Hong Guo , Ren Zhang , and Yuan Li1* 1 Key Laboratory of Biotechnology of Antibiotics, Ministry of Health, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Tiantan, Beijing 100050, China 2 Institute of Pharmacology and Toxicology, 27 Taiping Road, Beijing 100850, China 3 School of Biological Sciences, University of Wollongong, NSW 2522, Australia Received: March 24, 2009 / Revised: April 18, 2009 / Accepted: April 28, 2009 A novel exopolysaccharide named Ebosin was produced by donor and play significant roles in important biological Streptomyces sp. 139, with medicinal activity. Its biosynthesis processes [6]. gene cluster (ste) has been previously identified. For the Many bacteria are known to produce exopolysaccharides functional study of the ste7 gene in Ebosin biosynthesis, it (EPSs), which are excreted into the environment. The was disrupted with a double crossover via homologous biosynthesis of EPSs that consist of repeating units includes recombination. The monosaccharide composition of EPS- their assembly on a lipid carrier by sequential transfer of 7m produced by the mutant strain Streptomyces sp. 139 monosaccharides from nucleotide sugars by glycosyltransferases - (ste7 ) was found altered from that of Ebosin, with fucose (GTFs) and the subsequent polymerization and export of decreasing remarkably. For biochemical characterization of the repeating units [24].