Microvolume Chemical Analysis of Atmospheric Gases and Aerosols
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CARI-7 Documentation: Radiation Transport in the Atmosphere
DOT/FAA/AM-21/5 Office of Aerospace Medicine Washington, DC 20591 CARI Documentation: Radiation Transport in the Atmosphere Kyle Copeland Civil Aerospace Medical Institute Federal Aviation Administration Oklahoma City, OK 73125Location/Address March 2021 Final Report NOTICE This document is disseminated under the sponsorship of the U.S. Department of Transportation in the interest of information exchange. The United States Government assumes no liability for the contents thereof. _________________ This publication and all Office of Aerospace Medicine technical reports are available in full-text from the Civil Aerospace Medical Institute’s publications Web site: (www.faa.gov/go/oamtechreports) Technical Report Documentation Page 1. Report No. 2. Government Accession No. 3. Recipient's Catalog No. DOT/FAA/AM-21/5 4. Title and Subtitle 5. Report Date CARI-7 DOCUMENTATION: RADIATION TRANSPORT IN March 2021 THE ATMOSPHERE 6. Performing Organization Code 7. Author(s) 8. Performing Organization Report No. Copeland, K. 9. Performing Organization Name and Address 10. Work Unit No. (TRAIS) Civil Aerospace Medical Institute FAA 11. Contract or Grant No. 12. Sponsoring Agency name and Address 13. Type of Report and Period Covered Office of Aerospace Medicine Federal Aviation Administration 800 Independence Ave., S.W. Washington, DC 20591 14. Sponsoring Agency Code 15. Supplemental Notes 16. Abstract Primary cosmic radiation from both the Sun and interstellar space enters Earth's atmosphere in varying amounts. Outside of Earth's atmosphere, cosmic radiation is modulated by solar activity and Earth's magnetic field. Once the radiation enters Earth's atmosphere, it interacts with Earth's atmosphere in the same manner regardless of its point of origin (solar or galactic). -

Elemental Geosystems, 5E (Christopherson) Chapter 2 Solar Energy, Seasons, and the Atmosphere
Elemental Geosystems, 5e (Christopherson) Chapter 2 Solar Energy, Seasons, and the Atmosphere 1) Our planet and our lives are powered by A) energy derived from inside Earth. B) radiant energy from the Sun. C) utilities and oil companies. D) shorter wavelengths of gamma rays, X-rays, and ultraviolet. Answer: B 2) Which of the following is true? A) The Sun is the largest star in the Milky Way Galaxy. B) The Milky Way is part of our Solar System. C) The Sun produces energy through fusion processes. D) The Sun is also a planet. Answer: C 3) Which of the following is true about the Milky Way galaxy in which we live? A) It is a spiral-shaped galaxy. B) It is one of millions of galaxies in the universe. C) It contains approximately 400 billion stars. D) All of the above are true. E) Only A and B are true. Answer: D 4) The planetesimal hypothesis pertains to the formation of the A) universe. B) galaxy. C) planets. D) ocean basins. Answer: C 5) The flattened structure of the Milky Way is revealed by A) the constellations of the Zodiac. B) a narrow band of hazy light that stretches across the night sky. C) the alignment of the planets in the solar system. D) the plane of the ecliptic. Answer: B 6) Earth and the Sun formed specifically from A) the galaxy. B) unknown origins. C) a nebula of dust and gases. D) other planets. Answer: C 7) Which of the following is not true of stars? A) They form in great clouds of gas and dust known as nebula. -

Today: Upper Atmosphere/Ionosphere
Today: Upper Atmosphere/Ionosphere • Review atmospheric Layers – Follow the energy! – What heats the Stratosphere? • Mesosphere – Most turbulent layer – why? • Ionosphere – What is a Plasma? The Solar Spectrum: The amount of energy the Sun produces at a given wavelength is determined by its temperature. This is a general property of any Black Body such as a star or the heating element on the stove. • The Sun is about 5270 K and produces most light in the visible. • The atmosphere is transparent in the visible and so over half of the solar energy reaches the ground. • Some gasses absorb certain wavelengths of sunlight, and thus some energy is absorbed directly into the atmosphere. Depositing Energy Energy from the Sun may be transmitted directly to the ground, absorbed in the atmosphere, or reflected from clouds or the Sun back into space. Some re-radiated heat from the ground is absorbed by the atmosphere, further heating it. The Troposhphere The troposphere is the region of the atmosphere we live in. The primary source of energy in the troposphere is heat (infrared light ) radiated from the ground. This means that it is warmest at the bottom and coolest at the top. Temperature drops about 11.5 ° F for each km of altitude. When pressure and temperature (with temp. faster) drop with altitude it triggers convection . Convection makes the troposphere unstable, but in a good way. Dominant Region The Troposphere contains 80% of the mass in the atmosphere and 99% of the water in the atmosphere. Water Vapor, CO 2, methane, nitrous oxide, ozone, chloroflorocarbons are all greenhouse gases Convection (both horizontal and vertical) produces weather in the atmosphere. -

The Earth's Atmosphere
A Resource Booklet for SACE Stage 1 Earth and Environmental Science The following pages have been prepared by practicing teachers of SACE Earth and Environmental Science. The six Chapters are aligned with the six topics described in the SACE Stage 1 subject outline. They aim to provide an additional source of contexts and ideas to help teachers plan to teach this subject. For further information, including the general and assessment requirements of the course see: https://www.sace.sa.edu.au/web/earth-and-environmental- science/stage-1/planning-to-teach/subject-outline A Note for Teachers The resources in this booklet are not intended for ‘publication’. They are ‘drafts’ that have been developed by teachers for teachers. They can be freely used for educational purposes, including course design, topic and lesson planning. Each Chapter is a living document, intended for continuous improvement in the future. Teachers of Earth and Environmental Science are invited to provide feedback, particularly suggestions of new contexts, field-work and practical investigations that have been found to work well with students. Your suggestions for improvement would be greatly appreciated and should be directed to our project coordinator:: [email protected] Preparation of this booklet has been coordinated and funded by the Geoscience Pathways Project, under the sponsorship of the Geological Society of Australia (GSA) and the Teacher Earth Science Education Program (TESEP). In-kind support has been provided by the SA Department of Energy and Mining (DEM) and the Geological Survey of South Australia (GSSA). FAIR USE: The teachers named alongside each chapter of this document have researched available resources, selected and collated these notes and images from a wide range of sources. -
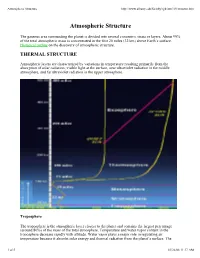
Atmospheric Structure
Atmospheric Structure http://www.albany.edu/faculty/rgk/atm101/structur.htm Atmospheric Structure The gaseous area surrounding the planet is divided into several concentric strata or layers. About 99% of the total atmospheric mass is concentrated in the first 20 miles (32 km) above Earth's surface. Historical outline on the discovery of atmospheric structure. THERMAL STRUCTURE Atmospheric layers are characterized by variations in temperature resulting primarily from the absorption of solar radiation; visible light at the surface, near ultraviolet radiation in the middle atmosphere, and far ultraviolet radiation in the upper atmosphere. Troposphere The troposphere is the atmospheric layer closest to the planet and contains the largest percentage (around 80%) of the mass of the total atmosphere. Temperature and water vapor content in the troposphere decrease rapidly with altitude. Water vapor plays a major role in regulating air temperature because it absorbs solar energy and thermal radiation from the planet's surface. The 1 of 5 01/26/08 11:17 AM Atmospheric Structure http://www.albany.edu/faculty/rgk/atm101/structur.htm troposphere contains 99 % of the water vapor in the atmosphere. Water vapor concentrations vary with latitude. They are greatest above the tropics, where they may be as high as 3 %, and decrease toward the polar regions. All weather phenomena occur within the troposphere, although turbulence may extend into the lower portion of the stratosphere. Troposphere means "region of mixing" and is so named because of vigorous convective air currents within the layer. The upper boundary of the layer, known as the tropopause, ranges in height from 5 miles (8 km) near the poles up to 11 miles (18 km) above the equator. -

The Earth's Atmosphere
TheThe EaEartrthh’s’s AtAtmomosphsphereere Earth’Earth’ss AtmospAtmospheherree AtmospAtmospherehere isis thethe gaseougaseouss layerlayer thatthat surrsurroundsounds thethe eeartharth IsIs aa mixmixtureture ofof ggasesases tthathat isis nanaturturallallyy odorodorlleess,ss, colourcolourless,less, tastetastelessless andand forformlessmless AiAirr isis blendblendeded soso thorthoroughlyoughly tthathat itit behavebehavess asas ifif itit wweerere aa singlesingle ggasas ThThee AtAtmospmospheherere AiAirr isis heldheld toto tthehe earearthth bbyy thethe forceforce ofof ggravity:ravity: TThehe furfurtherther awayaway frfromom thethe ssoourceurce ofof ggravitatravitatiiononalal attrattractionaction (e(eartharth)) ththee lowerlower thethe aattrattractictioonalnal forforcece MoMorere airair molecmoleculesules araree heldheld clclososerer toto thethe earearthth ththanan atat highehigherr elevaelevatitioonsns AtmosphAtmosphereere isis mormoree dedensense nenearar ththee sursurfaceface tthanhan atat highhigherer elevatioelevationsns ThThee AtAtmospmospheherere NoNo rerealal “t“top”op” –– atmatmospherospheree ddriftsrifts ofofff toto nothingnothingnessness aabovebove aboaboutut 101000 kmkm 97%97% ofof tthehe weighweightt andand 100%100% ofof tthehe watewaterr vapovaporr reresisiddee iinn thethe bottbottomom 3300 kmkm DDensiensittyy TThhee dendensisittyy (m(massass perper ununiitt volume)volume) ofof thethe atmoatmossppherehere quicklyquickly decrdecreaseseases wwithith iinncreacreassiningg elevatelevatiioonn aboaboveve seasea llevevelel DensityDensity -

1. Atmospheric Basics
Copyright © 2017 by Roland Stull. Practical Meteorology: An Algebra-based Survey of Atmospheric Science. v1.02 1 ATMOSPHERIC BASICS Contents Classical Newtonian physics can be used to de- scribe atmospheric behavior. Namely, air motions 1.1. Introduction 1 obey Newton’s laws of dynamics. Heat satisfies the 1.2. Meteorological Conventions 2 laws of thermodynamics. Air mass and moisture 1.3. Earth Frameworks Reviewed 3 are conserved. When applied to a fluid such as air, 1.3.1. Cartography 4 these physical processes describe fluid mechanics. 1.3.2. Azimuth, Zenith, & Elevation Angles 4 Meteorology is the study of the fluid mechanics, 1.3.3. Time Zones 5 physics, and chemistry of Earth’s atmosphere. 1.4. Thermodynamic State 6 The atmosphere is a complex fluid system — a 1.4.1. Temperature 6 system that generates the chaotic motions we call 1.4.2. Pressure 7 weather. This complexity is caused by myriad in- 1.4.3. Density 10 teractions between many physical processes acting 1.5. Atmospheric Structure 11 at different locations. For example, temperature 1.5.1. Standard Atmosphere 11 differences create pressure differences that drive 1.5.2. Layers of the Atmosphere 13 winds. Winds move water vapor about. Water va- 1.5.3. Atmospheric Boundary Layer 13 por condenses and releases heat, altering the tem- 1.6. Equation of State– Ideal Gas Law 14 perature differences. Such feedbacks are nonlinear, 1.7. Hydrostatic Equilibrium 15 and contribute to the complexity. 1.8. Hypsometric Equation 17 But the result of this chaos and complexity is a fascinating array of weather phenomena — phe- 1.9. -

The Problem of Using Mariner Iv Ionospheric Densities to Deduce a Model of the Martian Atmospheric Structure
i- . 8 -’ I 8 SPACE RESEARCH COORDI - ----’ --“T=D 1 GPO PRICE $- $- CFSTI PRICE(S) $ / Microfiche (MF) , Ls ff 853 July 65 THE PROBLEM OF USING MARINER IV IONOSPHERIC DENSITIES TO DEDUCE A MODEL OF THE MARTIAN ATMOSPHERIC STRUCTURE BY T. M. DONAHUE DEPARTMENT OF PHYSICS SRCC REPORT NO. 41 UNIVERSITY OF PITTSBURGH PITTSBURGH, PENNSYLVANIA 7 APRIL 1967 THE PROBLEM OF USING MARINER IV IONOSPHERIC DENSITIES TO DEDUCE A MODEL OF THE MARTIAN ATMOSPHERIC STRUCTURE T. M. Donahue University of Pittsburgh Pittsburgh, Pennsylvania April 1967 Reproduction in whole or in part is permissible for any purpose of the United States Government. The Problem of Using Mariner IV Ionospheric Densities to Deduce a Model of the Martian Atmospheric Structure (Presented at the Conference on The Atmospheres of Mars and Venus Kitt Peak National Iaboratory, February, 1967) T. M. Donahue University of Pittsburgh ABSTRACT An F1 layer maximum would apparently occur fifteen kilometers too high in the Martian ionosphere even though the atmosphere were pure C02 in a model with high temperature mesopause and thermopause. It is pointed out, however, that a similar calculation of electron densities in the earth's ionosphere starting with a reasonable model for neutral structure and temperature would also disagree with the observed profile of electron density in the F1 region. It is argued that where no ionospheric model based on unforced assumptions manifestly supports any atmospheric model it is unreasonable to rely on ionospheric properties to discriminate between models. Recently I suggested(') that the Martian ionosphere observed by Mariner IV(2) might be an F1 layer if the temperature profile was close to that calcuhted by Chamberlain and M~Elroy(~)and the lower atmosphere was almost pure C02 with a smface pressure of 6 mb. -

Atmospheric Processes and Weather We Have Looked at Water Flowing Over the Land in Considerable Detail
Atmospheric Processes and Weather We have looked at water flowing over the land in considerable detail. Another key component in the hydrologic cycle is the atmosphere and we will now look at in more detail at the atmosphere and the role water and air play in atmospheric processes and the hazards and benefits of these processes. Structure of Earth’s Atmosphere The diagram below shows the various layers that exist in the Earth’s atmosphere, their typical height above the Earth and the typical temperature in these layers. The air pressure drops with height above the ground as the air thins. Closest to the ground is the troposphere. This is the layer we spend most of our time living in, and in which the Earth’s weather occurs, including horizontal winds. The temperature drops with increasing height in the troposphere. The troposphere is about 18 km deep above the equator, but only 8 km deep above the Earth’s Poles. Near the top of the troposphere is the jet stream – strong, generally westerly winds in a narrow layer. The tropopause is the boundary between the troposphere and the stratosphere. Air temperature increases with height in the stratosphere. The stratosphere is almost weatherless, extends up to 50 km and contains the ozonosphere. The ozonosphere, or ozone layer, contains ozone, a form of oxygen which protects life on Earth by screening out harmful UV rays from the Sun. Without the ozone layer, life on Earth would struggle to survive, especially on land. The stratopause is the boundary between the stratosphere and the mesosphere. -

As200-C2l1:Lq1) A
The Atmosphere 5 dangers? A. Expert - I have done a lot of learning in this 0 area already. B. Above average - I have learned some 0 information about this topic. C. Moderate - I know a little bit about this topic. 0 D. Rookie - I am a blank slate...but I'm ready to 0 learn! (AS200-C2L1:LQ1) A. Stratosphere 1 B. Ozone 1 C. Emissions 1 D. Atmosphere 1 (AS200-C2L1:LQ2) Chapter Overview Lesson 1: The Atmosphere Lesson 2: Weather Elements Lesson 3: Aviation Weather Lesson 4: Weather Forecasting Lesson 5: The Effects of Weather on Aircraft Chapter 2, Lesson 1 Lesson Overview The atmosphere’s regions The roles of water and particulate matter in the atmosphere The primary causes of atmospheric motion The types of clouds How the atmospheric layers impact flight Chapter 2, Lesson 1 Click any link below to go directly to polling that question. 1. Person who forecasts weather 8. Amount of water in atmosphere 2. Blanket of air, surrounds Earth 9. Actual amount of moisture in 3. Strong air current, flows west to air compared with total amount east of moisture air could hold at 4. Transformation, liquid to gas that temperature 5. Solid to gas, bypass liquid state 10. Temperature at which air can 6. Water state change, gas-liquid hold no more moisture Click here to return to this index. Click any link below to go directly to polling that question. 11. As full of moisture as something 16. Line parallel to Earth’s equator can get 17. Height above Earth’s surface of 12. -

Modeling Earth's Atmospheric Layers
Student Sheet 1 CRITICAL THINKING ACTIVITY: MODELING EARTH’S ATMOSPHERIC LAYERS The Earth is surrounded by the atmosphere, a body of air or gasses that protects the planet and enables life to exist. Most of our atmosphere is located close to the earth's surface where it is most dense. The air of our planet is 79% nitrogen and just under 21% oxygen; the small amount remaining is composed of carbon dioxide and other gases. There are five distinct layers of the Earth’s atmosphere: Troposphere: The layer of the atmosphere closest to the Earth is the troposphere. This layer is where weather occurs. It begins at the surface of the earth and extends out to about 4- 12 miles. The temperature of the troposphere decreases with height. This layer is known as the lower atmosphere. Stratosphere: Above the troposphere is the stratosphere, which extends to about 30-35 miles above the earth's surface. Temperature rises within the stratosphere but still remains well below freezing. Jet streams occur here, which are fast moving currents of air between the 2 layers. 1 Student Sheet 2 Mesosphere: From about 35 to 50 miles above the surface of the earth lies the mesosphere, where the air is especially thin and molecules are great distances apart. Temperatures in the mesosphere reach a low of -184°F (-120°C). It is the coldest layer of the atmosphere. Radio waves are reflected to Earth and meteors burn up in this layer. The stratosphere and the mesosphere are the middle atmosphere. Thermosphere: The thermosphere rises several hundred miles above the earth's surface, from 50 miles up to about 400 miles. -

Earth's Atmosphere: the Thermosphere And
EARTH’S ATMOSPHERE: THE THERMOSPHERE AND MESOPAUSE Among the 1000 km (600 miles) and 500 km (300 miles) of altitude below the exosphere is the “thermopause” or “exobase” on top of the “thermosphere” and its altitude varies with the solar activity. In this area there are ions of hydrogen and helium, while below this layer there are heavier gases as monoatomic oxygen. From this area you begin to perceive the atmospheric pressure, so you can say that this is the ends the “outer space”. On top of “Thermosphere”, between 600 km (400 miles) and 200 km (120 miles) of altitude there is an accumulation of the denser layer of the “ionosphere” which include nitrogen, oxygen, ionized hydrogen and helium atoms and electrons which diffuse into layers according to their density where the lighter particles are in the upper zones, occurring convections due to changes in heat. During the day the “ionosphere” layer intercepts high energy photons, strongly interacting with ultraviolet radiation of between 10 and 100 nm, X-rays and short radio waves, being highly dependent on the solar weather activity. The shocks generate heat vibrations reaching 1500 °K at 400 km (250 miles) of altitude and the frequent combination of ions and electrons, causing auroras and electrical currents. Also in the ionosphere meteorites disintegrate resulting in shooting stars. From the 160 km (100 miles) of altitude, particles begin to be close enough to spread sound, while at 120 km (75 miles) of altitude the effects of atmospheric entry of an object from outer space start to be felt, although they contain lower density than the thermopause which has temperatures of about 700 ° K.