Anticholinergic Substances
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent Office
3,092,548 United States Patent Office Patented June 4, 1963 2 are the various side effects that occur in an appreciable 3,092,548 number of patients, the foremost of which are a blurring METHOD OF TREATING PEPTICULCERS WITH PANTOTHENECACD of vision, drowsiness and a general dry condition mani Albert G. Worton, Columbus, Ohio, assignor to The fested by a retarded salivation, reduction of perspiration Warren-Teed Products Company, Columbus, Ohio, a 5 and diminished urinary output. Other side effects which corporation of Ohio occur in some cases are glaucoma, stimulation of the cen No Drawing. Filed Oct. 27, 1960, Ser. No. 65,256 trol nervous system and in severe cases cardiac and respi 5 Claims. (Ci. 67-55) ratory collapse. These side effects increase to Some de gree with the increase in dosage. Despite the side effects This invention relates to preparation adapted for use O these compounds are fairly selective and highly effective in treating disorders of the gastro-intestinal tract, more in decreasing the volume and acidity of gastric secretion particularly in treating peptic ulcer, both of the duodenal and in reducing gastrointestinal motility. and gastric type, for hyperacidity, hypertropic gastritis, Understandably, the main problem in the past has been splenic flexure syndrome, biliary dyskinesia (postchole to provide a dosage of anticholinergic drug which will cystectomy syndrome) and hiatal hernia or other condi achieve the most beneficial results possible and yet mini tions where anticholinergic effect, either spasmolytic or mize the undesired side effects. One of the objects of antisecretory is indicated or where antiuicerogenic effect this invention is to provide novel compositions containing is indicated. -
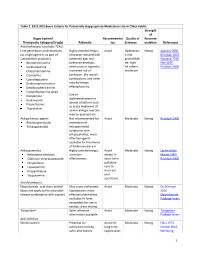
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Isopropamide Iodide
www.chemicalland21.com ISOPROPAMIDE IODIDE SYNONYMS (3-Carbamoyl-3,3-diphenylpropyl)diisopropylmethylammonium iodide; 2,2-Diphenyl-4- diisopropylaminobutyramide methiodide; 4-(Diisopropylamino)-2,2-diphenylbutyramide methiodide; gamma-(Aminocarbonyl)-N-methyl-N,N-bis(1-methylethyl)-gamma-phenylbenzenepropanaminium iodide; Iodure d'isopropamide; Ioduro de isopropamida; Isopropamide ioduro; Isopropamidi iodidum; Isoproponum iodide; PRODUCT IDENTIFICATION CAS RN 71-81-8 EINECS RN 200-766-8 FORMULA C23H33IN2O MOL WEIGHT 480.43 PHYSICAL AND CHEMICAL PROPERTIES PHYSICAL STATE white to off-white powder MELTING POINT 199 C BOILING POINT DENSITY SOLUBILITY IN WATER pH VAPOR DENSITY REFRACTIVE INDEX FLASH POINT GENERAL DESCRIPTION Isopropamide is a long-acting anticholinergic and antimuscarinic drug of quaternary ammonium structure. It is used in the form of the iodide, (also bromide or chloride) to treat peptic ulcer and to suppress gastric secretion other gastrointestinal disorders. Brands of Isopropamide drugs: Darbid Dipramide Isamide Marygin-M Piaccamide Priamide Priazimide Sanulcin Tyrimide Quaternary ammonium anticholinergics (Synthetic) ATC Code Product CAS RN. A03AB01 Benzilonium bromide 1050-48-2 A03AB02 Glycopyrrolate 596-51-0 A03AB03 Oxyphenonium 14214-84-7 A03AB04 Penthienate 22064-27-3 A03AB05 Propantheline 50-34-0 A03AB06 Otilonium bromide 26095-59-0 A03AB07 Methantheline 5818-17-7 Please mail us if you want to sell your product or need to buy some products) www.chemicalland21.com ISOPROPAMIDE IODIDE A03AB08 Tridihexethyl 60-49-1 A03AB09 Isopropamide 7492-32-2 A03AB10 Hexocyclium 6004-98-4 A03AB11 Poldine 596-50-9 A03AB12 Mepenzolic acid 25990-43-6 A03AB13 Bevonium 33371-53-8 A03AB14 Pipenzolate 13473-38-6 A03AB15 Diphemanil methylsulfate 62-97-5 A03AB16 (2-Benzhydryloxyethyl)diethyl-methylammonium iodide A03AB17 Tiemonium iodide 144-12-7 A03AB18 Prifinium bromide 4630-95-9 A03AB19 Timepidium bromide 35035-05-3 A03AB21 Fenpiverinium bromide 125-60-0 03AB53 Oxyphenonium, combinations STABILITY AND REACTIVITY STABILITY Stable under normal conditions. -

The National Drugs List
^ ^ ^ ^ ^[ ^ The National Drugs List Of Syrian Arab Republic Sexth Edition 2006 ! " # "$ % &'() " # * +$, -. / & 0 /+12 3 4" 5 "$ . "$ 67"5,) 0 " /! !2 4? @ % 88 9 3: " # "$ ;+<=2 – G# H H2 I) – 6( – 65 : A B C "5 : , D )* . J!* HK"3 H"$ T ) 4 B K<) +$ LMA N O 3 4P<B &Q / RS ) H< C4VH /430 / 1988 V W* < C A GQ ") 4V / 1000 / C4VH /820 / 2001 V XX K<# C ,V /500 / 1992 V "!X V /946 / 2004 V Z < C V /914 / 2003 V ) < ] +$, [2 / ,) @# @ S%Q2 J"= [ &<\ @ +$ LMA 1 O \ . S X '( ^ & M_ `AB @ &' 3 4" + @ V= 4 )\ " : N " # "$ 6 ) G" 3Q + a C G /<"B d3: C K7 e , fM 4 Q b"$ " < $\ c"7: 5) G . HHH3Q J # Hg ' V"h 6< G* H5 !" # $%" & $' ,* ( )* + 2 ا اوا ادو +% 5 j 2 i1 6 B J' 6<X " 6"[ i2 "$ "< * i3 10 6 i4 11 6! ^ i5 13 6<X "!# * i6 15 7 G!, 6 - k 24"$d dl ?K V *4V h 63[46 ' i8 19 Adl 20 "( 2 i9 20 G Q) 6 i10 20 a 6 m[, 6 i11 21 ?K V $n i12 21 "% * i13 23 b+ 6 i14 23 oe C * i15 24 !, 2 6\ i16 25 C V pq * i17 26 ( S 6) 1, ++ &"r i19 3 +% 27 G 6 ""% i19 28 ^ Ks 2 i20 31 % Ks 2 i21 32 s * i22 35 " " * i23 37 "$ * i24 38 6" i25 39 V t h Gu* v!* 2 i26 39 ( 2 i27 40 B w< Ks 2 i28 40 d C &"r i29 42 "' 6 i30 42 " * i31 42 ":< * i32 5 ./ 0" -33 4 : ANAESTHETICS $ 1 2 -1 :GENERAL ANAESTHETICS AND OXYGEN 4 $1 2 2- ATRACURIUM BESYLATE DROPERIDOL ETHER FENTANYL HALOTHANE ISOFLURANE KETAMINE HCL NITROUS OXIDE OXYGEN PROPOFOL REMIFENTANIL SEVOFLURANE SUFENTANIL THIOPENTAL :LOCAL ANAESTHETICS !67$1 2 -5 AMYLEINE HCL=AMYLOCAINE ARTICAINE BENZOCAINE BUPIVACAINE CINCHOCAINE LIDOCAINE MEPIVACAINE OXETHAZAINE PRAMOXINE PRILOCAINE PREOPERATIVE MEDICATION & SEDATION FOR 9*: ;< " 2 -8 : : SHORT -TERM PROCEDURES ATROPINE DIAZEPAM INJ. -

United States Patent (19) 11 4,111,203 Theeuwes (45) "Sep
United States Patent (19) 11 4,111,203 Theeuwes (45) "Sep. 5, 1978 54) OSMOTIC SYSTEM WITH MEANS FOR Primary Examiner-Benjamin R. Padgett MPROVING DELVERY KNETCS OF Assistant Examiner-T. S. Gron SYSTEM Attorney, Agent, or Firm-Paul L. Sabatine; Thomas E. 75 Inventor: Felix Theeuwes, Los Altos, Calif. Ciotti; Edward L. Mandell 73) Assignee: Alza Corporation, Palo Alto, Calif. 57 ABSTRACT * Notice: The portion of the term of this patent An osmotic system for delivering a beneficial agent is subsequent to Sep. 5, 1995, has been disclosed. The system comprises a wall surrounding a disclaimed. compartment and has a passageway through the wall for delivering agent from the compartment. The wall is (21) Appl. No.: 744,089 formed of a material permeable to the passage of an (22 Filed: Nov. 22, 1976 external fluid and impermeable to the passage of agent. 51) Int. Cl’............................................. A61M31/00 The compartment contains an agent that is soluble in 52 U.S. C. ..................................... 128/260; 206/0.5; the fluid and exhibits an osmotic pressure gradient 222/130; 222/193; 222/389; 222/395; 222/491; across the wall against the fluid, or the compartment 424/19 contains an agent that has limited solubility in the fluid 58) Field of Search ............... 128/260, 261,268, 272; and exhibits a limited osmotic pressure gradient across 424/15, 19-22, 33, 37; 222/491,395, 193,389, the wall against the fluid. The compartment also con 130; 206/0.5 tains means for increasing the amount of agent deliv ered from the system. The means comprises a film sur (56) References Cited rounding an osmagent with the film formed of a mate U.S. -

Muscarinic Acetylcholine Receptor
mAChR Muscarinic acetylcholine receptor mAChRs (muscarinic acetylcholine receptors) are acetylcholine receptors that form G protein-receptor complexes in the cell membranes of certainneurons and other cells. They play several roles, including acting as the main end-receptor stimulated by acetylcholine released from postganglionic fibersin the parasympathetic nervous system. mAChRs are named as such because they are more sensitive to muscarine than to nicotine. Their counterparts are nicotinic acetylcholine receptors (nAChRs), receptor ion channels that are also important in the autonomic nervous system. Many drugs and other substances (for example pilocarpineand scopolamine) manipulate these two distinct receptors by acting as selective agonists or antagonists. Acetylcholine (ACh) is a neurotransmitter found extensively in the brain and the autonomic ganglia. www.MedChemExpress.com 1 mAChR Inhibitors & Modulators (+)-Cevimeline hydrochloride hemihydrate (-)-Cevimeline hydrochloride hemihydrate Cat. No.: HY-76772A Cat. No.: HY-76772B Bioactivity: Cevimeline hydrochloride hemihydrate, a novel muscarinic Bioactivity: Cevimeline hydrochloride hemihydrate, a novel muscarinic receptor agonist, is a candidate therapeutic drug for receptor agonist, is a candidate therapeutic drug for xerostomia in Sjogren's syndrome. IC50 value: Target: mAChR xerostomia in Sjogren's syndrome. IC50 value: Target: mAChR The general pharmacol. properties of this drug on the The general pharmacol. properties of this drug on the gastrointestinal, urinary, and reproductive systems and other… gastrointestinal, urinary, and reproductive systems and other… Purity: >98% Purity: >98% Clinical Data: No Development Reported Clinical Data: No Development Reported Size: 10mM x 1mL in DMSO, Size: 10mM x 1mL in DMSO, 1 mg, 5 mg 1 mg, 5 mg AC260584 Aclidinium Bromide Cat. No.: HY-100336 (LAS 34273; LAS-W 330) Cat. -

Drugs to Avoid in Patients with Dementia
Detail-Document #240510 -This Detail-Document accompanies the related article published in- PHARMACIST’S LETTER / PRESCRIBER’S LETTER May 2008 ~ Volume 24 ~ Number 240510 Drugs To Avoid in Patients with Dementia Elderly people with dementia often tolerate drugs less favorably than healthy older adults. Reasons include increased sensitivity to certain side effects, difficulty with adhering to drug regimens, and decreased ability to recognize and report adverse events. Elderly adults with dementia are also more prone than healthy older persons to develop drug-induced cognitive impairment.1 Medications with strong anticholinergic (AC) side effects, such as sedating antihistamines, are well- known for causing acute cognitive impairment in people with dementia.1-3 Anticholinergic-like effects, such as urinary retention and dry mouth, have also been identified in drugs not typically associated with major AC side effects (e.g., narcotics, benzodiazepines).3 These drugs are also important causes of acute confusional states. Factors that may determine whether a patient will develop cognitive impairment when exposed to ACs include: 1) total AC load (determined by number of AC drugs and dose of agents utilized), 2) baseline cognitive function, and 3) individual patient pharmacodynamic and pharmacokinetic features (e.g., renal/hepatic function).1 Evidence suggests that impairment of cholinergic transmission plays a key role in the development of Alzheimer’s dementia. Thus, the development of the cholinesterase inhibitors (CIs). When used appropriately, the CIs (donepezil [Aricept], rivastigmine [Exelon], and galantamine [Razadyne, Reminyl in Canada]) may slow the decline of cognitive and functional impairment in people with dementia. In order to achieve maximum therapeutic effect, they ideally should not be used in combination with ACs, agents known to have an opposing mechanism of action.1,2 Roe et al studied AC use in 836 elderly patients.1 Use of ACs was found to be greater in patients with probable dementia than healthy older adults (33% vs. -

NINDS Custom Collection II
ACACETIN ACEBUTOLOL HYDROCHLORIDE ACECLIDINE HYDROCHLORIDE ACEMETACIN ACETAMINOPHEN ACETAMINOSALOL ACETANILIDE ACETARSOL ACETAZOLAMIDE ACETOHYDROXAMIC ACID ACETRIAZOIC ACID ACETYL TYROSINE ETHYL ESTER ACETYLCARNITINE ACETYLCHOLINE ACETYLCYSTEINE ACETYLGLUCOSAMINE ACETYLGLUTAMIC ACID ACETYL-L-LEUCINE ACETYLPHENYLALANINE ACETYLSEROTONIN ACETYLTRYPTOPHAN ACEXAMIC ACID ACIVICIN ACLACINOMYCIN A1 ACONITINE ACRIFLAVINIUM HYDROCHLORIDE ACRISORCIN ACTINONIN ACYCLOVIR ADENOSINE PHOSPHATE ADENOSINE ADRENALINE BITARTRATE AESCULIN AJMALINE AKLAVINE HYDROCHLORIDE ALANYL-dl-LEUCINE ALANYL-dl-PHENYLALANINE ALAPROCLATE ALBENDAZOLE ALBUTEROL ALEXIDINE HYDROCHLORIDE ALLANTOIN ALLOPURINOL ALMOTRIPTAN ALOIN ALPRENOLOL ALTRETAMINE ALVERINE CITRATE AMANTADINE HYDROCHLORIDE AMBROXOL HYDROCHLORIDE AMCINONIDE AMIKACIN SULFATE AMILORIDE HYDROCHLORIDE 3-AMINOBENZAMIDE gamma-AMINOBUTYRIC ACID AMINOCAPROIC ACID N- (2-AMINOETHYL)-4-CHLOROBENZAMIDE (RO-16-6491) AMINOGLUTETHIMIDE AMINOHIPPURIC ACID AMINOHYDROXYBUTYRIC ACID AMINOLEVULINIC ACID HYDROCHLORIDE AMINOPHENAZONE 3-AMINOPROPANESULPHONIC ACID AMINOPYRIDINE 9-AMINO-1,2,3,4-TETRAHYDROACRIDINE HYDROCHLORIDE AMINOTHIAZOLE AMIODARONE HYDROCHLORIDE AMIPRILOSE AMITRIPTYLINE HYDROCHLORIDE AMLODIPINE BESYLATE AMODIAQUINE DIHYDROCHLORIDE AMOXEPINE AMOXICILLIN AMPICILLIN SODIUM AMPROLIUM AMRINONE AMYGDALIN ANABASAMINE HYDROCHLORIDE ANABASINE HYDROCHLORIDE ANCITABINE HYDROCHLORIDE ANDROSTERONE SODIUM SULFATE ANIRACETAM ANISINDIONE ANISODAMINE ANISOMYCIN ANTAZOLINE PHOSPHATE ANTHRALIN ANTIMYCIN A (A1 shown) ANTIPYRINE APHYLLIC -

Pharmacy and Poisons (Third and Fourth Schedule Amendment) Order 2017
Q UO N T FA R U T A F E BERMUDA PHARMACY AND POISONS (THIRD AND FOURTH SCHEDULE AMENDMENT) ORDER 2017 BR 111 / 2017 The Minister responsible for health, in exercise of the power conferred by section 48A(1) of the Pharmacy and Poisons Act 1979, makes the following Order: Citation 1 This Order may be cited as the Pharmacy and Poisons (Third and Fourth Schedule Amendment) Order 2017. Repeals and replaces the Third and Fourth Schedule of the Pharmacy and Poisons Act 1979 2 The Third and Fourth Schedules to the Pharmacy and Poisons Act 1979 are repealed and replaced with— “THIRD SCHEDULE (Sections 25(6); 27(1))) DRUGS OBTAINABLE ONLY ON PRESCRIPTION EXCEPT WHERE SPECIFIED IN THE FOURTH SCHEDULE (PART I AND PART II) Note: The following annotations used in this Schedule have the following meanings: md (maximum dose) i.e. the maximum quantity of the substance contained in the amount of a medicinal product which is recommended to be taken or administered at any one time. 1 PHARMACY AND POISONS (THIRD AND FOURTH SCHEDULE AMENDMENT) ORDER 2017 mdd (maximum daily dose) i.e. the maximum quantity of the substance that is contained in the amount of a medicinal product which is recommended to be taken or administered in any period of 24 hours. mg milligram ms (maximum strength) i.e. either or, if so specified, both of the following: (a) the maximum quantity of the substance by weight or volume that is contained in the dosage unit of a medicinal product; or (b) the maximum percentage of the substance contained in a medicinal product calculated in terms of w/w, w/v, v/w, or v/v, as appropriate. -

Wo 2010/075090 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 1 July 2010 (01.07.2010) WO 2010/075090 A2 (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C07D 409/14 (2006.01) A61K 31/7028 (2006.01) kind of national protection available): AE, AG, AL, AM, C07D 409/12 (2006.01) A61P 11/06 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (21) International Application Number: DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/US2009/068073 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, 15 December 2009 (15.12.2009) ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (25) Filing Language: English SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, (26) Publication Language: English TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 61/122,478 15 December 2008 (15.12.2008) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, (71) Applicant (for all designated States except US): AUS- ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, PEX PHARMACEUTICALS, INC. -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -

Microgram Journal, Vol 2, Number 1
Washington, D. C. Office of Science and Education Vol.II,No.1 Division of Laboratory Operations January 1969 INDEXISSUE CORRECTION 11 "Structure Elucidation of 'LBJ' , by Sander W. Bellman, John W. Turczan, James Heagy and Ted M. Hopes, Micro Gram .!., 3, 6-13 (Dec. 1968) Page 7, third and fourth sentences under Discussion: Change to read: "The melting point of the acid moiety found in step (g) was 148-150°c., compared to the litera ture, v~lue of 151°c for the melting point of benzilic acid (2); thus the benzilic acid melting point gives support to the proposed structure for 'LBJ'. Spectral evidence also supports the proposed structure". MICRO-GRAMREVISION Please re-number the pages of your copies of Micro-Gram, Volume I. Re-number pages bearing printing only. Vol ume I will then be numbered from page 1, the front page of issue No. 1, through page 189 the last page of issue No. 12. To help with this task, pages contained within each issue are as follows: Issue Number Page Through 1 1 8 2 9 29 3 30 32 4 33 66 5 67 79 6 80 97 7 98 120 8 121 128 9 129 136 10 137 157 11 158 170 12 171 189 CAUTION: Use of this publication should be restricted to forensic analysts or others having a legitimate need for this material. From the Archive Library of Erowid Center http://erowid.org/library/periodicals/microgram -2- CANNABIS ,·,-...__/' Attached is a copy of 11A Short Rapid Method for the Identification of Cannabis." The method was developed by Mro H.D.