Tissue-Engineered Models for Glaucoma Research
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Experimental Glaucoma Model with Controllable Intraocular Pressure History Kayla R
www.nature.com/scientificreports OPEN Experimental glaucoma model with controllable intraocular pressure history Kayla R. Ficarrotta1, Youssef H. Mohamed1 & Christopher L. Passaglia1,2* Glaucoma-like neuropathies can be experimentally induced by disturbing aqueous outfow from the eye, resulting in intraocular pressure (IOP) changes that are variable in magnitude and time course and permanent in duration. This study introduces a novel method of glaucoma induction that ofers researchers round-the-clock measurement and reversible control of IOP for the frst time. One eye of Brown-Norway rats was implanted with a cannula tethered to a pressure sensor and aqueous reservoir. IOP was raised 10 mmHg for weeks-to-months in treated animals and unaltered in control animals. Counts of Brn3a-expressing retinal ganglion cells (RGCs) in implanted eyes were indistinguishable from non-implanted eyes in control animals and 15 ± 2%, 23 ± 4%, and 38 ± 4% lower in animals exposed to 2, 4, and 9 weeks of IOP elevation. RGC loss was greater in peripheral retina at 2 weeks and widespread at longer durations. Optic nerves also showed progressive degeneration with exposure duration, yet conventional outfow facility of implanted eyes was normal (24.1 ± 2.9 nl/min/mmHg) even after 9-weeks elevation. Hence, this infusion-based glaucoma model exhibits graded neural damage with unimpaired outfow pathways. The model further revealed a potentially-signifcant fnding that outfow properties of rat eyes do not remodel in response to chronic ocular hypertension. Glaucoma is a heterogeneous group of ocular disorders characterized by progressive and preferential loss of ret- inal ganglion cells (RGCs), resulting in visual feld defcits and ultimately blindness. -

Japanese Journal of Ophthalmology Vol.43 No.4
Autonomic Nerves Containing Substance P in the Aqueous Outflow Channels and Scleral Spur of the Guinea Pig Mariko Sasamoto, Hai-Bo Chen and Shigeo Tsukahara Department of Ophthalmology, Yamanashi Medical University, Tamaho, Yamanashi, Japan Purpose: To study the innervation of the aqueous outflow channels and scleral spur by auto- nomic nerves containing substance P. Methods: The experiments were conducted on guinea pigs. Immunohistochemical tech- niques and capsaicin-ablation of the sensory nerves were used to investigate nerves contain- ing substance P at the light and electron microscopic level. Results: Nerves containing substance P were observed in the aqueous outflow channels and scleral spur regions. The fine structures of these nerves had a similar pattern in those regions, and the labeled elements had abundant small vesicles, a few large vesicles, and numerous neurotubuli. Following capsaicin treatment, these nerves remained intact and no degener- ated substance P-like immunoreactive nerves were found. Conclusions: Nerves containing substance P are most likely of autonomic origin in view of their ultrastructural features. These nerves innervate the aqueous outflow channels and scleral spur, and are probably important for neurogenic influences on the intraocular pres- sure by the autonomic nervous system. Jpn J Ophthalmol 1999;43:272–278 © 1999 Japa- nese Ophthalmological Society Key Words: Aqueous outflow channels, capsaicin, guinea pig, immunohistochemistry, substance P. Introduction The scleral spur region should also be considered 4,5 Aqueous outflow channels and the scleral spur because it plays a part in outflow regulation. The human scleral spur contains elastic tissue, similar to play important roles in the regulation of intraocular sclera, so that changes in the IOP might affect these pressure (IOP), and are known to have varied pepti- 5 dergic innervation, which have received special at- tissues. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

The Narrow Range of Intraocular Pressure (TOP) (12-20Mmhg)
J. Smooth Muscle Res. 32: 229•`247, 1996. Review Ocular Outflow Facility with Emphasis on Neuronal Regulation of Intraocular Smooth Muscles Ryo SUZUKI, MD Department of Ophthalmology, Yamaguchi University School of Medicine, Ube City, 755, Japan The narrow range of intraocular pressure (TOP) (12-20mmHg) in normal individuals has stimulated a search for possible regulatory mechanisms7,18,65) of aqueous production and outflow6,46). Compared with aqueous production, the aqueous outflow mechanisms of the neuronal, humoral, and mechanical processes have been studied much less. Because the eye constitutes a small portion of total body mass, it is very difficult to determine the neuronal and mechanical regulations of intraocular muscle and outflow facility. Locally acting mechanisms, should be an ideal means for integrating its physiology58,76). The peripheral nervous system is designed to effect such local control. Because most available antiglaucoma agents interact with the autonomic mechanisms and mechanical activities of the smooth muscles in the eye53,59,68),combined studies of the intraocular muscles with eye perfusion7,66) and cell shape changes of cultured cells from the outflow route22,66) would suggest the role of the nervous system in regulating IOP. From an historical view point, much speculation and minimal experimentation have been focused on the influence of the iris sphincter, the iris dilator, and the ciliary muscles on aqueous humor outflow. Accommodation, cholinergic agonists, and stimulation of the oculomotor nerve, all increase outflow facility19), whereas ganglionic blocking agents and anticholinergic drugs decrease the ocular outflow facility5,66). Furthermore, the outflow facility increase with intravenous pilocarpine administration is instantaneous, suggesting that the effect is mediated by an arterially perfused tissue31,32,46). -

Simultaneous Influence of Sympathetic Autonomic Stress on Schlemm's
www.nature.com/scientificreports OPEN Simultaneous infuence of sympathetic autonomic stress on Schlemm’s canal, intraocular pressure and ocular circulation Wei Chen, Zhiqi Chen, Yan Xiang, Chaohua Deng, Hong Zhang & Junming Wang* This study aimed to investigate changes in Schlemm’s canal, intraocular pressure and ocular blood circulation following the activation of the sympathetic nervous system. Twenty healthy volunteers were enrolled in this study. The cold pressor test (CPT) was adopted. Cross-sectional area of Schlemm’s canal (SCAR), superfcial and deep retinal vessel densities (s-RVD;d-RVD), pupil diameter (PD), intraocular pressure (IOP), mean ocular perfusion pressure (MOPP) and heart rate variability (HRV) were measured at three time-points: baseline (T0) and 5 min (T1) and 10 min (T2) after the CPT. After cold stimulation, LF/HF index (the ratio of low frenquency and high frenquency) increased signifcantly. IOP decreased from 16.9 ± 1.9 mmHg at baseline to 16.4 ± 2.7 mmHg at T1 and to 15.2 ± 2.7 mmHg at T2. The nasal cross-sectional area of SCAR (SCAR-n) increased from 6283.9 ± 2696.2 µm2 at baseline to 8392.9 ± 3258.7 µm2 at T1 and to 10422.0 ± 3643.8 µm2 at T2. The temporal cross-sectional area of SCAR (SCAR-t) increased from 6414.5 ± 2218.7 µm2 at baseline to 8610.8 ± 2317.1 µm2 at T1 and to 11544.0 ± 4129.2 µm2 at T2. The expansion of Schlemm’s canal was observed after the CPT might be caused by sympathetic nerve stimulation, subsequently leading to decreased IOP. -
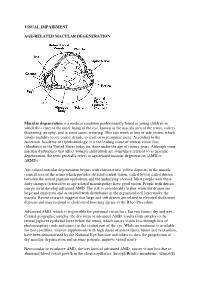
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye. -

1 42. Ophthalmology Daniel G Vaughan Ocular Emergencies It Is
42. Ophthalmology Daniel G Vaughan Ocular emergencies It is not necessary to refer every patient with an eye disease to an ophthalmologist for treatment. In general, sties, bacterial conjunctivitis, superficial trauma to the lids, corneas, and conjunctiva, and superficial corneal foreign bodies can be treated just as effectively by the surgeon or primary physician as by the ophthalmologist. More serious eye disease such as the following should be referred as soon as possible for specialized care: iritis, acute glaucoma, retinal detachment, strabismus, contusion of the globe, and severe corneal trauma or infection. In the management of acute ocular disorders, it is most important to establish a definitive diagnosis before prescribing treatment. The maxim "All red eyes are not pinkeye" is a useful one, and the physician must be alert for the more serious iritis, keratitis, or glaucoma. The common practice of prescribing "shotgun" topical antibiotic combinations containing corticosteroids is to be discouraged, because inappropriate use of steroids can lead to complications. This chapter attempts to summarize the basic principles and technics of diagnosis and management of common ocular problems, with special emphasis on emergencies, particularly those caused by trauma. Ocular emergencies may be classified as true emergencies or urgent cases. A true emergency is one in which the patient is suffering severe pain or in which a few hours' delay in treatment can lead to permanent ocular damage. An urgent case is one in which treatment should be started as soon as possible but in which a delay of a few days can be tolerated. Foreign Bodies If a patient complains of "something in my eye" and gives a consistent history, a foreign body is usually present even though it may not be readily visible. -

Human Trabecular Mesh Work Organ Culture: Morphology and Glycosaminoglycan Synthesis
Investigative Ophthalmology & Visual Science, Vol. 29, No. 1, January 1988 Copyright © Association for Research in Vision and Ophthalmology Human Trabecular Mesh work Organ Culture: Morphology and Glycosaminoglycan Synthesis Ted 5. Acott,* Paul D. Kingsley, John R. Samples, and E. Michael Van Buskirk Human corneoscleral explants were maintained for several weeks in defined, serum-free media. Tra- becular cell vitality, as judged by vital stain exclusion, is high for at least one month. Trabecular ultrastructure, as compared to that of fresh eyes, first shows minor cellular and extracellular matrix degradation after 3 weeks in culture. The biosynthetic profiles of trabecular glycosaminoglycans (GAGs) change significantly by 3 weeks in culture. Eyes that are stored at 5°C for up to 48 hr postmortem exhibit changes in trabecular ultrastructure and in GAG profiles; both characteristics return to normal by 7 days in culture. The incorporation pattern of 35S-sulfate and 3H-glucosamine into the GAGs of the trabecular meshwork (TM) is distinct from corneal or scleral incorporation. The relative incorporation of 3H-glucosamine into trabecular GAGs, as determined by sequential enzy- matic degradation, is: 22.3% hyaluronic acid (HA), 27.9% chondroitin sulfate (CS), 21.3% dermatan sulfate (DS), 5.9% keratan sulfate (KS), 17.7% heparan sulfate (HS) and 4.9% unidentified material. The relative incorporation of 35S-sulfate into trabecular GAGs is: 0% HA, 32.9% CS, 34.8% DS, 7.7% KS, 13.8% HS and 11.1% into unidentified material. This profile is in good agreement with the profile that was previously obtained for human and nonhuman primate meshworks prior to culture. -

The Development of the Trabecular Meshwork and Its Abnormality in Primary- Infantile Glaucoma*
THE DEVELOPMENT OF THE TRABECULAR MESHWORK AND ITS ABNORMALITY IN PRIMARY- INFANTILE GLAUCOMA* BY Douglas R. Anderson, MD INTRODUCTION IN CONGENITAL GLAUCOMA, THE PERIPHERAL IRIS IS SEEN BY GONIOSCOPY TO BE inserted more anteriorly than normal, and the trabecular meshwork seems to have a shiny surface. 1-4 At the time of goniotomy, this surface tissue is incised, and the peripheral iris falls posteriorly as if the incised tissue had been holding the iris insertion forward. Since the operation usually relieves the elevated pressure,4 it is natural to assume that the shiny surface is an imperforate membrane that had prevented aqueous humor outflow. Histopathologic confirmation of this presumed membrane or other pathologic features typical of infantile glaucoma has been difficult to obtain. Specimens enucleated after long-standing severe glaucoma, per- haps after several surgical procedures, contain secondary changes that hide the nature ofthe initial abnormality. Other infantile glaucoma speci- mens were obtained at autopsy from infants who died of associated fatal congenital anomalies. The abnormalities in these eyes may not be the same as the abnormality that produces primary infantile glaucoma in otherwise normal infants. Furthermore, most investigators have not clari- fied which morphologic features of the angles of infantile glaucomatous eyes are characteristics of infant eyes (known to differ from adult eyes *From the Bascom Palmer Eye Institute, Department of Ophthalmology, University of Miami School of Medicine, Miami, Florida. This investigation was supported in part by Public Health Service Research Grant EY-00031 from the National Eye Institute, Bethesda, Maryland, by the Glaucoma Re- search Fund of the American Health Assistance Foundation, by the John Russell Stubbins Foundation, and by the Phillips Foundation. -

Targeting the Trabecular Meshwork, Schlemm's Canal and the Collector Channels
WHITEPAPER Targeting the Trabecular Meshwork, Schlemm’s Canal and the Collector Channels The Evolution of Canaloplasty and the iTrack™ Canaloplasty Microcatheter As our understanding of the pathogenesis of glaucoma has evolved, the role of the proximal versus distal portions of the conventional outflow pathway as therapeutic targets has generated growing interest. It is well understood that the trabecular meshwork, specifically the extracellular matrix filling the open space of the juxtacanalicular connective tissue (JCT) is the primary site of outflow resistance in the conventional outflow pathway.1 It is also known that glaucomatous pathology can cause significant outflow resistance in distal pathways.2 Greater contractility of Schlemm’s canal has been shown to increase outflow resistance.3,4,5 Further, the canal in eyes with primary open angle glaucoma (POAG) tends to be shorter, narrowed, and often collapsed, reducing the area of active flow.3,4,5 Another significant cause of increased outflow resistance in POAG eyes is herniations of the trabecular meshwork obstructing up to 90% of collector channels.6 Armed with improved Introduced in 2005 and approved First conceived through an understanding of the function by the FDA in 2008, canaloplasty ab-externo approach as an ™ and physiology of the proximal with the iTrack microcatheter alternative to trabeculectomy, has evolved to be a procedure surgeons have developed and distal outflow pathways in with high utility in the treatment an ab-interno canaloplasty glaucomatous eyes, an increasing of glaucoma. It is indicated in a technique that is conjunctiva number of surgeons are turning wide variety of glaucoma types and sclera sparing and thus to ab-interno canaloplasty to including; POAG, pigmentary can be deployed earlier in the target each segment of the (PG), pseudoexfoliation (PXF) disease process. -

Regenerative Capacity of the Corneal Transition Zone for Endothelial Cell Therapy
Sie et al. Stem Cell Research & Therapy (2020) 11:523 https://doi.org/10.1186/s13287-020-02046-2 REVIEW Open Access Regenerative capacity of the corneal transition zone for endothelial cell therapy Nicole Ming Sie1,2, Gary Hin-Fai Yam1,3* , Yu Qiang Soh1,2, Matthew Lovatt1, Deepinder Dhaliwal3, Viridiana Kocaba1,4 and Jodhbir S. Mehta1,2,5,6* Abstract The corneal endothelium located on the posterior corneal surface is responsible for regulating stromal hydration. This is contributed by a monolayer of corneal endothelial cells (CECs), which are metabolically active in a continuous fluid-coupled efflux of ions from the corneal stroma into the aqueous humor, preventing stromal over- hydration and preserving the orderly arrangement of stromal collagen fibrils, which is essential for corneal transparency. Mature CECs do not have regenerative capacity and cell loss due to aging and diseases results in irreversible stromal edema and a loss of corneal clarity. The current gold standard of treatment for this worldwide blindness caused by corneal endothelial failure is the corneal transplantation using cadaveric donor corneas. The top indication is Fuchs corneal endothelial dystrophy/degeneration, which represents 39% of all corneal transplants performed. However, the global shortage of transplantable donor corneas has restricted the treatment outcomes, hence instigating a need to research for alternative therapies. One such avenue is the CEC regeneration from endothelial progenitors, which have been identified in the peripheral endothelium and the adjacent transition zone. This review examines the evidence supporting the existence of endothelial progenitors in the posterior limbus and summarizes the existing knowledge on the microanatomy of the transitional zone. -

Guidelines for the Collaborative Management of Persons with Age-Related Macular Degeneration by Health- and Eye-Care Professionals C Clinical Research
CANADIAN JOURNAL of OPTOMETRY | REVUE CANadienne d’OPTOMÉTRIE EST. 1939 VOLUME 77 ISSUE 1 DIGITAL Guidelines for the Collaborative Management of Persons with Age-Related Macular Degeneration by Health- and Eye-Care Professionals C CLINICAL RESEARCH Guidelines for the Collaborative Management of Persons with Age-Related Macular Degeneration by Health- and Eye-Care Professionals BACKGROUND Age-related macular degeneration (AMD) is the most common cause of severe visual impair- ment in adults in the developed world. Statistics released by the CNIB report that AMD now represents more than 50% of new referrals to their organization.1 Considered in light of the growing incidence and prevalence of this disease and an aging population, AMD poses a major public health challenge. As its name suggests, AMD is an age-related degenerative disease affecting approximately 1.5% of Canadians by age 40 years, rising to affect a full 25% of society by age 75. It begins with the development of drusen, yellowish deposits of waste material within the macula. This gradu- ally progresses with advancing age and may result in the loss of central (detail) vision. Such progression ultimately affects a person’s ability to read, drive, and perform many other activities of daily living.2 AMD is typically divided into two forms: 1. Non-exudative or dry AMD, which affects approximately 85% of patients with AMD, but typically progresses slowly and causes less visual impairment. 2. Exudative or wet AMD affects approximately 15% of patients with AMD, but is responsible for greater visual impairment that can occur within weeks or months of development.