Ap2s1 Mutation in Mice Causes Familial Hypocalciuric Hypercalcemia Type 3
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Sarcoidosis, Hypercalcemia and Calcium-Sensing Receptor Mutation: a Case Report
468 Letters to the Editor Sarcoidosis, Hypercalcemia and Calcium-sensing Receptor Mutation: A Case Report Shreya Dixit1 and Pablo Fernandez-Peñas1,2* 1Skin and Cancer Foundation Australia, 7 Ashley Lane, Westmead, NSW 2145, and 2Sydney Medical School Western, The University of Sydney, NSW, Australia. *E-mail: [email protected] Accepted October 28, 2010. Sarcoidosis is a multisystem granulomatous disease of The incidence of hypercalcaemia in patients with unknown aetiology. It predominantly affects the lungs; sarcoidosis is approximately 10% (3). It has previously however, 25% of patients have skin involvement, ranging been found that these abnormalities of calcium metabo- from non-specific maculopapular eruptions to erythema lism are due to dysregulated production of calcitriol by nodosum and lupus pernio (1). activated macrophages trapped in pulmonary alveoli and Familial hypocalciuric hypercalcaemia (FHH) results granulomatous inflammation (4). Conversion of calcidiol from an autosomal dominantly inherited inactivating to calcitriol is facilitated by 1 alpha-hydroxylase, a cyto- mutation of the calcium-sensing receptor (CaSR) gene chrome P450 enzyme. The activity of 1 alpha-hydroxyla- and is typically asymptomatic (2). Sarcoidosis has not se is tightly regulated through complex mechanisms that previously been reported in FHH. We had the opportu- depend on the circulating levels of calcium, phosphorus, nity to study a patient with sarcoidosis that has a CaSR PTH, 1,25 (OH)2D3 (calcitriol) and calcitonin. The role mutation. of CaSR in the regulation of 1 alpha-hydroxylase has not been defined, although there are suggestions that CaSR activation can repress the enzyme (5). CASE REPORT The incidence of FHH in patients with sarcoidosis A 70-year-old woman was referred by her endocrinologist with is unknown. -

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Phytoplankton As Key Mediators of the Biological Carbon Pump: Their Responses to a Changing Climate
sustainability Review Phytoplankton as Key Mediators of the Biological Carbon Pump: Their Responses to a Changing Climate Samarpita Basu * ID and Katherine R. M. Mackey Earth System Science, University of California Irvine, Irvine, CA 92697, USA; [email protected] * Correspondence: [email protected] Received: 7 January 2018; Accepted: 12 March 2018; Published: 19 March 2018 Abstract: The world’s oceans are a major sink for atmospheric carbon dioxide (CO2). The biological carbon pump plays a vital role in the net transfer of CO2 from the atmosphere to the oceans and then to the sediments, subsequently maintaining atmospheric CO2 at significantly lower levels than would be the case if it did not exist. The efficiency of the biological pump is a function of phytoplankton physiology and community structure, which are in turn governed by the physical and chemical conditions of the ocean. However, only a few studies have focused on the importance of phytoplankton community structure to the biological pump. Because global change is expected to influence carbon and nutrient availability, temperature and light (via stratification), an improved understanding of how phytoplankton community size structure will respond in the future is required to gain insight into the biological pump and the ability of the ocean to act as a long-term sink for atmospheric CO2. This review article aims to explore the potential impacts of predicted changes in global temperature and the carbonate system on phytoplankton cell size, species and elemental composition, so as to shed light on the ability of the biological pump to sequester carbon in the future ocean. -
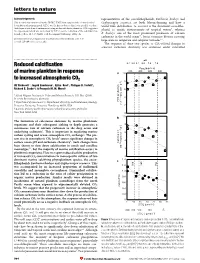
Reduced Calcification of Marine Plankton in Response to Increased
letters to nature Acknowledgements representatives of the coccolithophorids, Emiliania huxleyi and This research was sponsored by the EPSRC. T.W.F. ®rst suggested the electrochemical Gephyrocapsa oceanica, are both bloom-forming and have a deoxidation of titanium metal. G.Z.C. was the ®rst to observe that it was possible to reduce world-wide distribution. G. oceanica is the dominant coccolitho- thick layers of oxide on titanium metal using molten salt electrochemistry. D.J.F. suggested phorid in neritic environments of tropical waters9, whereas the experiment, which was carried out by G.Z.C., on the reduction of the solid titanium dioxide pellets. M. S. P. Shaffer took the original SEM image of Fig. 4a. E. huxleyi, one of the most prominent producers of calcium carbonate in the world ocean10, forms extensive blooms covering Correspondence and requests for materials should be addressed to D. J. F. large areas in temperate and subpolar latitudes9,11. (e-mail: [email protected]). The response of these two species to CO2-related changes in seawater carbonate chemistry was examined under controlled ................................................................. pH Reduced calci®cation 8.4 8.2 8.1 8.0 7.9 7.8 PCO2 (p.p.m.v.) of marine plankton in response 200 400 600 800 a 10 to increased atmospheric CO2 ) 8 –1 Ulf Riebesell *, Ingrid Zondervan*, BjoÈrn Rost*, Philippe D. Tortell², d –1 Richard E. Zeebe*³ & FrancËois M. M. Morel² 6 * Alfred Wegener Institute for Polar and Marine Research, P.O. Box 120161, 4 D-27515 Bremerhaven, Germany mol C cell –13 ² Department of Geosciences & Department of Ecology and Evolutionary Biology, POC production Princeton University, Princeton, New Jersey 08544, USA (10 2 ³ Lamont-Doherty Earth Observatory, Columbia University, Palisades, New York 10964, USA 0 ............................................................................................................................................. -

Distribution of Calcium Phosphate in the Exoskeleton of Larval Exeretonevra Angustifrons Hardy (Diptera: Xylophagidae)
Arthropod Structure & Development 34 (2005) 41–48 www.elsevier.com/locate/asd Distribution of calcium phosphate in the exoskeleton of larval Exeretonevra angustifrons Hardy (Diptera: Xylophagidae) Bronwen W. Cribba,b,*, Ron Rascha, John Barrya, Christopher M. Palmerb,1 aCentre for Microscopy and Microanalysis, The University of Queensland, Brisbane, Qd 4072, Australia bDepartment of Zoology and Entomology, The University of Queensland, Brisbane, Qd 4072, Australia Received 28 July 2004; accepted 26 August 2004 Abstract Distribution and organisation of the mineral, amorphous calcium phosphate (ACP), has been investigated in the exoskeleton of the xylophagid fly larva Exeretonevra angustifrons Hardy. While head capsule and anal plate are smooth with a thin epicuticle, the epicuticle of the body is thicker and shows unusual micro-architecture comprised of minute hemispherical (dome-shaped) protrusions. Electron microprobe analysis and energy dispersive spectroscopy revealed heterogeneity of mineral elements across body cuticle and a concentration of ACP in the epicuticle, especially associated with the hemispherical structures. Further imaging and analysis showed the bulk of the ACP to be present in nano-sized granules. It is hypothesised that the specific distribution of ACP may enhance cuticular hardness or durability without reducing flexibility. q 2004 Elsevier Ltd. All rights reserved. Keywords: Insect; Cuticle; Integument; Hardening; Analytical electron microscopy; Electron microprobe 1. Introduction was distributed heterogeneously. Further investigation of the distribution of the mineral phase at the micron and Strengthening of biological structures through cuticular nanometre level is needed to discover where deposition is calcification is well developed in decapod crustaceans but it occurring and how this might affect exoskeletal organis- rarely occurs in insects, where it is poorly understood ation. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Effects of Ocean Acidification and Sea-Level Rise on Coral Reefs
Effects of Ocean Acidification and Sea-Level Rise on Coral Reefs Coral reefs are vital to the long-term to produce CaCO3, carbon dioxide (CO2), As CO2 increases in the atmosphere, viability of coastal societies, providing and water (H2O). Over time as these more is absorbed by the surface of the economic, recreational, and aesthetic organisms grow and die, their skeletons ocean, where it combines with seawater value from which coastal communities break down and become calcium carbon- to make a weak acid called carbonic thrive. Some of the services that coral ate sediments. These sediments fill in acid (H2CO3). This process, called reefs provide include protection from the framework of the reef and eventually ocean acidification, causes a decrease storm waves, nurseries and habitats for become cemented together, construct- in seawater pH (or increase in acidity) commercially important fish species, ing the foundation for continued upward that can result in a decrease in biogenic and production of sand for beaches. growth of the reef structure. The infilling calcification rates, dissolution of carbon- Coral reefs develop over thousands of the reef framework with sediments is ate sediments, and loss of reef structure. of years as tropical marine organ- what allows vertical accretion over time One of the primary concerns associated isms build skeletons of calcium carbonate and enables reef growth to keep up with with ocean acidification is whether coral (CaCO3) minerals to form a three-dimen- sea-level rise. Calcification is a revers- reefs will be able to continue to grow at sional structure (fig. 1). This process, ible process. -

Table S2.Up Or Down Regulated Genes in Tcof1 Knockdown Neuroblastoma N1E-115 Cells Involved in Differentbiological Process Anal
Table S2.Up or down regulated genes in Tcof1 knockdown neuroblastoma N1E-115 cells involved in differentbiological process analysed by DAVID database Pop Pop Fold Term PValue Genes Bonferroni Benjamini FDR Hits Total Enrichment GO:0044257~cellular protein catabolic 2.77E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 537 13588 1.944851 8.64E-07 8.64E-07 5.02E-07 process ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, USP17L5, FBXO11, RAD23B, NEDD8, UBE2V2, RFFL, CDC GO:0051603~proteolysis involved in 4.52E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 534 13588 1.93519 1.41E-06 7.04E-07 8.18E-07 cellular protein catabolic process ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, USP17L5, FBXO11, RAD23B, NEDD8, UBE2V2, RFFL, CDC GO:0044265~cellular macromolecule 6.09E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 609 13588 1.859332 1.90E-06 6.32E-07 1.10E-06 catabolic process ISG15, RBM8A, ATG7, LOC100046898, PSENEN, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, XRN2, USP17L5, FBXO11, RAD23B, UBE2V2, NED GO:0030163~protein catabolic process 1.81E-09 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 556 13588 1.87839 5.64E-06 1.41E-06 3.27E-06 ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, -

Ykt6 Membrane-To-Cytosol Cycling Regulates Exosomal Wnt Secretion
bioRxiv preprint doi: https://doi.org/10.1101/485565; this version posted December 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Ykt6 membrane-to-cytosol cycling regulates exosomal Wnt secretion Karen Linnemannstöns1,2, Pradhipa Karuna1,2, Leonie Witte1,2, Jeanette Kittel1,2, Adi Danieli1,2, Denise Müller1,2, Lena Nitsch1,2, Mona Honemann-Capito1,2, Ferdinand Grawe3,4, Andreas Wodarz3,4 and Julia Christina Gross1,2* Affiliations: 1Hematology and Oncology, University Medical Center Goettingen, Goettingen, Germany. 2Developmental Biochemistry, University Medical Center Goettingen, Goettingen, Germany. 3Molecular Cell Biology, Institute I for Anatomy, University of Cologne Medical School, Cologne, Germany 4Cluster of Excellence-Cellular Stress Response in Aging-Associated Diseases (CECAD), Cologne, Germany *Correspondence: Dr. Julia Christina Gross, Hematology and Oncology/Developmental Biochemistry, University Medical Center Goettingen, Justus-von-Liebig Weg 11, 37077 Goettingen Germany Abstract Protein trafficking in the secretory pathway, for example the secretion of Wnt proteins, requires tight regulation. These ligands activate Wnt signaling pathways and are crucially involved in development and disease. Wnt is transported to the plasma membrane by its cargo receptor Evi, where Wnt/Evi complexes are endocytosed and sorted onto exosomes for long-range secretion. However, the trafficking steps within the endosomal compartment are not fully understood. The promiscuous SNARE Ykt6 folds into an auto-inhibiting conformation in the cytosol, but a portion associates with membranes by its farnesylated and palmitoylated C-terminus. Here, we demonstrate that membrane detachment of Ykt6 is essential for exosomal Wnt secretion. -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g. -

Biomineralization and Evolutionary History Andrew H
1 111 Biomineralization and Evolutionary History Andrew H. Knoll Department of Organismic and Evolutionary Biology Harvard University Cambridge, Massachusetts, 02138 U.S.A. INTRODUCTION The Dutch ethologist Niko Tinbergen famously distinguished between proximal and ultimate explanations in biology. Proximally, biologists seek a mechanistic understanding of how organisms function; most of this volume addresses the molecular and physiological bases of biomineralization. But while much of biology might be viewed as a particularly interesting form of chemistry, it is more than that. Biology is chemistry with a history, requiring that proximal explanations be grounded in ultimate, or evolutionary, understanding. The physiological pathways by which organisms precipitate skeletal minerals and the forms and functions of the skeletons they fashion have been shaped by natural selection through geologic time, and all have constrained continuing evolution in skeleton-forming clades. In this chapter, I outline some major patterns of skeletal evolution inferred from phylogeny and fossils (Figure 1), highlighting ways that our improving mechanistic knowledge of biomineralization can help us to understand this evolutionary record (see Leadbetter and Riding 1986; Lowenstam and Weiner 1989; Carter 1990; and Simkiss and Wilbur 1989 for earlier reviews). Figure 1. A geologic time scale for the past 1000 million years, showing the principal time divisions used in Earth science and the timing of major evolutionary events discussed in this chapter. Earlier intervals of time—the Mesoproterozoic (1600–1000 million years ago) and Paleoproterozoic (2500– 1600 million years ago) eras of the Proterozoic Eon and the Archean Eon (> 2500 million years ago)— are not shown. Time scale after Remane (2000). -

A Novel Pharmacodynamic Assay to Evaluate the Effects of Crystallization
www.nature.com/scientificreports OPEN A novel pharmacodynamic assay to evaluate the efects of crystallization inhibitors on calcium Received: 1 March 2017 Accepted: 26 June 2017 phosphate crystallization in human Published online: 31 July 2017 plasma M. D. Ferrer1, M. M. Pérez1, M. M. Cànaves1, J. M. Buades 2, C. Salcedo1 & J. Perelló1,3 Cardiovascular calcifcation (CVC) is a progressive complication of chronic kidney disease and a predictor of CV events and mortality. The use of biomarkers to predict CV risk and activities of potential or current treatment drugs in these patients could have a crucial impact on therapeutic approaches. Our aim was to develop a novel assay for measurement of the rate of calcium phosphate crystallization in human plasma and provide a tool to evaluate the efects of crystallization inhibitors. The efcacy of inhibitors was determined by adding inhibitory compounds (polyphosphates, fetuin-A, sodium thiosulfate or citrate) to control samples. The assay was additionally validated for SNF472, an experimental formulation of phytate being developed for the treatment of calciphylaxis and CVC in patients with end-stage renal disease (ESRD) undergoing hemodialysis (HD). The method was repeatable and reproducible. The plasma crystallization rate was reduced up to 80% in a concentration-dependent manner following treatment with inhibitors in vitro, among which SNF472 was the most potent. This method appears benefcial in evaluating and discriminating between inhibitory activities of compounds such as polyphosphates on calcium phosphate crystallization, which present a novel therapeutic approach to treat CVC in ESRD patients. Calcifcation is the normal process of calcium salt deposition in body tissues occurring due to the presence of supersaturated or metastable salt solutions in biological fuids1, 2.