Pauling's Conceptions of Hybridization and Resonance in Modern Quantum Chemistry
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Amino Acid Building Block Models – in Brief
Amino Acid Building Block Models – In Brief Key Teaching Points for Amino Acid Building Block Models© Overall Student Learning Objective: What Dictates How a Protein Folds? Amino acids are the building blocks of proteins. All amino acids have an identical core structure consisting of an alpha-carbon, carboxyl group, amino group and R-group (sidechain). A linear chain of amino acids is a polypeptide. The primary sequence of a protein is the linear sequence of amino acids in a polypeptide. Proteins are made up of amino acid monomers linked together by peptide bonds. Peptide bond formation between amino acids results in the release of water (dehydration synthesis or condensation reaction). The protein backbone is characterized by the “N-C-C-N-C-C. .” pattern. The “ends” of the protein can be identified by the N-terminus (amino group) end and the C-terminus (carboxyl group) end. For a more complete lesson guide, please visit: http://www.3dmoleculardesigns.com/3DMD-Files/AABB/ContentsandAssembly.pdf Amino Acid Core Structure Build an amino acid according to the diagram to the right: 1. Identify the alpha carbon, amino group, carboxyl group and R-group (sidechain representation) in the structure you have constructed. Two amino acids can be chemically linked by a reaction called “condensation” or “dehydration synthesis” to form a dipeptide bond linking the two amino acids. A chain of amino acid units (monomers) linked together by peptide bonds is called a polypeptide. General Dipeptide Structure Construct a model of a dipeptide using the amino acid models previously built. 2. What are the products of the condensation reaction (dehydration synthesis)? 3. -

Symmetry of Three-Center, Four-Electron Bonds†‡
Chemical Science EDGE ARTICLE View Article Online View Journal | View Issue Symmetry of three-center, four-electron bonds†‡ b a c Cite this: Chem. Sci., 2020, 11,7979 Ann Christin Reiersølmoen, § Stefano Battaglia, § Sigurd Øien-Ødegaard, Arvind Kumar Gupta, d Anne Fiksdahl,b Roland Lindh a and Mate Erdelyi *a All publication charges for this article ´ ´ ´ have been paid for by the Royal Society of Chemistry Three-center, four-electron bonds provide unusually strong interactions; however, their nature remains ununderstood. Investigations of the strength, symmetry and the covalent versus electrostatic character of three-center hydrogen bonds have vastly contributed to the understanding of chemical bonding, whereas the assessments of the analogous three-center halogen, chalcogen, tetrel and metallic s^-type long bonding are still lagging behind. Herein, we disclose the X-ray crystallographic, NMR spectroscopic and computational investigation of three-center, four-electron [D–X–D]+ bonding for a variety of cations (X+ ¼ H+,Li+,Na+,F+,Cl+,Br+,I+,Ag+ and Au+) using a benchmark bidentate model system. Formation of a three-center bond, [D–X–D]+ is accompanied by an at least 30% shortening of the D–X Received 11th April 2020 bonds. We introduce a numerical index that correlates symmetry to the ionic size and the electron Accepted 19th June 2020 affinity of the central cation, X+. Providing an improved understanding of the fundamental factors DOI: 10.1039/d0sc02076a Creative Commons Attribution 3.0 Unported Licence. determining bond symmetry on a comprehensive level is expected to facilitate future developments and rsc.li/chemical-science applications of secondary bonding and hypervalent chemistry. -

Introduction to Proteins and Amino Acids Introduction
Introduction to Proteins and Amino Acids Introduction • Twenty percent of the human body is made up of proteins. Proteins are the large, complex molecules that are critical for normal functioning of cells. • They are essential for the structure, function, and regulation of the body’s tissues and organs. • Proteins are made up of smaller units called amino acids, which are building blocks of proteins. They are attached to one another by peptide bonds forming a long chain of proteins. Amino acid structure and its classification • An amino acid contains both a carboxylic group and an amino group. Amino acids that have an amino group bonded directly to the alpha-carbon are referred to as alpha amino acids. • Every alpha amino acid has a carbon atom, called an alpha carbon, Cα ; bonded to a carboxylic acid, –COOH group; an amino, –NH2 group; a hydrogen atom; and an R group that is unique for every amino acid. Classification of amino acids • There are 20 amino acids. Based on the nature of their ‘R’ group, they are classified based on their polarity as: Classification based on essentiality: Essential amino acids are the amino acids which you need through your diet because your body cannot make them. Whereas non essential amino acids are the amino acids which are not an essential part of your diet because they can be synthesized by your body. Essential Non essential Histidine Alanine Isoleucine Arginine Leucine Aspargine Methionine Aspartate Phenyl alanine Cystine Threonine Glutamic acid Tryptophan Glycine Valine Ornithine Proline Serine Tyrosine Peptide bonds • Amino acids are linked together by ‘amide groups’ called peptide bonds. -

Polar Covalent Bonds: Electronegativity
Polar Covalent Bonds: Electronegativity Covalent bonds can have ionic character These are polar covalent bonds Bonding electrons attracted more strongly by one atom than by the other Electron distribution between atoms is not symmetrical Bond Polarity and Electronegativity Symmetrical Covalent Bonds Polar Covalent Bonds C – C + - C – H C – O (non-polar) (polar) Electronegativity (EN): intrinsic ability of an atom to attract the shared electrons in a covalent bond Inductive Effect: shifting of sigma bonded electrons in resppygonse to nearby electronegative atom The Periodic Table and Electronegativity C – H C - Br and C - I (non-pol)lar) (po l)lar) Bond Polarity and Inductive Effect Nonpolar Covalent Bonds: atoms with similar EN Polar Covalent Bonds: Difference in EN of atoms < 2 Ionic Bonds: Difference in EN > 2 C–H bonds, relatively nonpolar C-O, C-X bonds (more electronegative elements) are polar Bonding electrons shift toward electronegative atom C acqqppuires partial positive char g,ge, + Electronegative atom acquires partial negative charge, - Inductive effect: shifting of electrons in a bond in response to EN of nearby atoms Electrostatic Potential Maps Electrostatic potential maps show calculated charge distributions Colors indicate electron- rich (red) and electron- poor (blue ) reg ions Arrows indicate direction of bond polarity Polar Covalent Bonds: Net Dipole Moments Molecules as a whole are often polar from vector summation of individual bond polarities and lone-pair contributions Strongly polar substances soluble in polar solvents like water; nonpolar substances are insoluble in water. Dipole moment ( ) - Net molecular polarity, due to difference in summed charges - magnitude of charge Q at end of molecular dipole times distance r between charges = Q r, in debyes (D), 1 D = 3.336 1030 coulomb meter length of an average covalent bond, the dipole moment would be 1.60 1029 Cm, or 4.80 D. -

Heterocycles 2 Daniel Palleros
Heterocycles 2 Daniel Palleros Heterocycles 1. Structures 2. Aromaticity and Basicity 2.1 Pyrrole 2.2 Imidazole 2.3 Pyridine 2.4 Pyrimidine 2.5 Purine 3. Π-excessive and Π-deficient Heterocycles 4. Electrophilic Aromatic Substitution 5. Oxidation-Reduction 6. DNA and RNA Bases 7. Tautomers 8. H-bond Formation 9. Absorption of UV Radiation 10. Reactions and Mutations Heterocycles 3 Daniel Palleros Heterocycles Heterocycles are cyclic compounds in which one or more atoms of the ring are heteroatoms: O, N, S, P, etc. They are present in many biologically important molecules such as amino acids, nucleic acids and hormones. They are also indispensable components of pharmaceuticals and therapeutic drugs. Caffeine, sildenafil (the active ingredient in Viagra), acyclovir (an antiviral agent), clopidogrel (an antiplatelet agent) and nicotine, they all have heterocyclic systems. O CH3 N HN O O N O CH 3 N H3C N N HN N OH O S O H N N N 2 N O N N O CH3 N CH3 caffeine sildenafil acyclovir Cl S N CH3 N N H COOCH3 nicotine (S)-clopidogrel Here we will discuss the chemistry of this important group of compounds beginning with the simplest rings and continuing to more complex systems such as those present in nucleic acids. Heterocycles 4 Daniel Palleros 1. Structures Some of the most important heterocycles are shown below. Note that they have five or six-membered rings such as pyrrole and pyridine or polycyclic ring systems such as quinoline and purine. Imidazole, pyrimidine and purine play a very important role in the chemistry of nucleic acids and are highlighted. -
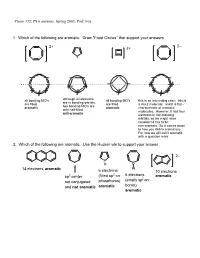
PS 6 Answers, Spring 2003, Prof
Chem 332, PS 6 answers, Spring 2003, Prof. Fox 1. Which of the following are aromatic. Draw "Frost Circles" that support your answers 2– 2+ 2+ + all bonding MO's although all electrons all bonding MO's this is an interesting case: this is are in bonding orbitals, are filled are filled a 4n+2 molecule, and it is flat-- two bonding MO's are aromatic aromatic characteristic of aromatic only half-filled molecules. However, it has four anti-aromatic electrons in non-bonding orbitals, so we might have considered this to be non-aromatic. So it comes down to how you define aromaticity. For now we will call it aromatic with a question mark. 2. Which of the following are aromatic. Use the Huckel rule to support your answer 2– P H B 14 electrons, aromatic H 6 electrons 10 electrons 2 6 electrons sp3 center (filled sp on aromatic (empty sp2 on not conjugated phosphorus) boron) and not aromatic aromatic aromatic 3. Compound 1 has an unusually large dipole moment for an organic hydrocarbon. Explain why (consider resonance structures for the central double bond) We need to remember that for any double bond compound, we can draw two polar resonance structure. Normally, these resonance structures are unimportant unimportant unimportant However, for 1 the story is different because there is a polar resonance form that has two aromatic components. In other words, 'breaking' the double bond gives rise to aromaticity, and therefore the polar resonance structure is important and leads to the observed dipole. dipole 1 8 e– 4 e– 6 e– 6 e– anti- anti- aromatic aromatic aromatic aromatic important resonance unimportant resonance structure structure 5. -

Structure of Benzene, Orbital Picture, Resonance in Benzene, Aromatic Characters, Huckel’S Rule B
Dr.Mrityunjay Banerjee, IPT, Salipur B PHARM 3rd SEMESTER BP301T PHARMACEUTICAL ORGANIC CHEMISTRY –II UNIT I UNIT I Benzene and its derivatives 10 Hours A. Analytical, synthetic and other evidences in the derivation of structure of benzene, Orbital picture, resonance in benzene, aromatic characters, Huckel’s rule B. Reactions of benzene - nitration, sulphonation, halogenationreactivity, Friedelcrafts alkylation- reactivity, limitations, Friedelcrafts acylation. C. Substituents, effect of substituents on reactivity and orientation of mono substituted benzene compounds towards electrophilic substitution reaction D. Structure and uses of DDT, Saccharin, BHC and Chloramine Benzene and its Derivatives Chemists have found it useful to divide all organic compounds into two broad classes: aliphatic compounds and aromatic compounds. The original meanings of the words "aliphatic" (fatty) and "aromatic" (fragrant/ pleasant smell). Aromatic compounds are benzene and compounds that resemble benzene in chemical behavior. Aromatic properties are those properties of benzene that distinguish it from aliphatic hydrocarbons. Benzene: A liquid that smells like gasoline Boils at 80°C & Freezes at 5.5°C It was formerly used to decaffeinate coffee and component of many consumer products, such as paint strippers, rubber cements, and home dry-cleaning spot removers. A precursor in the production of plastics (such as Styrofoam and nylon), drugs, detergents, synthetic rubber, pesticides, and dyes. It is used as a solvent in cleaning and maintaining printing equipment and for adhesives such as those used to attach soles to shoes. Benzene is a natural constituent of petroleum products, but because it is a known carcinogen, its use as an additive in gasoline is now limited. In 1970s it was associated with leukemia deaths. -

Introduction 1.1 Post-Translational Modifications
Chapter I: Introduction 1.1 Post-translational modifications Post-translational modifications (PTMs) are responsible for dynamic regulation of protein structure and function (Jensen et al., 2006). One example of the function of post-translational modifications in the dynamic regulation of protein structure and function is signal transduction; post-translational modification of key regulatory proteins in signal transduction pathways controls cellular proliferation, metabolism, gene expression, cytoskeletal organization and apoptosis (Jensen et al., 2006). Post-translational modifications not only change the mass of the protein, they are also often responsible for changes in charge, structure and conformation. This leads to changes in the biological activity of proteins like enzymes, the binding affinity of proteins to receptors and other protein-protein interactions (Hoffmann et al., 2008) (Murray et al., 1998). For example, the interaction of the cytokine TGF-beta with its receptor complex results in the phosphorylation of SMAD family proteins which activates these proteins to regulate target gene expression (Massague et al., 2000). Post-translational modified amino acid residues can act as binding sites on proteins for a specific recognition domain on other proteins. For example, phosphorylated tyrosine residues can bind to SH2 or PTB domains (Pawson et al., 1997). Depending on the type of PTM they can be very abundant and have a large number of target proteins, whereas other PTM`s have only a few target proteins. Moreover, some PTM`s are connected with several different residues and not only with one residue. For example multi-site phosphorylation that primarily targets Serine, Threonine and Tyrosine residues is a crucial mechanism for the regulation of protein localization and functional activity (Cohen et al., 2000). -

Structures and Reactivity of Transition-Metal Compounds
STRUCTURES AND REACTIVITY OF TRANSITION-METAL COMPOUNDS FEATURING METAL-LIGAND MULTIPLE BONDS A Dissertation by ZHENGGANG XU Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, Michael B. Hall Committee Members, Roland E. Allen Robert R. Lucchese Simon W. North Head of Department, David H. Russell August 2014 Major Subject: Chemistry Copyright 2014 Zhenggang Xu ABSTRACT This dissertation presents the results from density functional theory (DFT) calculations on three major projects I have been working on over the past several years. The first system is focused on the structure and reactivity of a novel osmium silylyne compounds featuring an Os≡Si triple bond. NMR simulation confirmed the existence of this compounds and bonding analysis like NBO and ETS-NOCV proved its triple bond character. The structures of the [2+2] cyclo-addition product from silylyne and other small molecules (PhC≡CPh and P≡CtBu) were also determined from possible isomers by energetic results and NMR simulations. The cycloaddition reactions were found to be under kinetic control and steric effects should be a major reason for that. Furthermore, the geometric and electronic structures of the osmium silylyne analogues (M≡E, M = Ru and Os; E = Si, Ge and Sn) are studied computationally and their similarities and distinctions are discussed. Both the second and third systems are related to the formation of transition metal imido compound (M=NR). In the second system a cationic oxorhenium(V) complex reacts with a series of arylazides (N3Ar) to give cationic cis-rhenium(VII) oxo imido complexes. -

Peptides & Proteins
Peptides & Proteins (thanks to Hans Börner) 1 Proteins & Peptides Proteuos: Proteus (Gr. mythological figure who could change form) proteuo: „"first, ref. the basic constituents of all living cells” peptos: „Cooked referring to digestion” Proteins essential for: Structure, metabolism & cell functions 2 Bacterium Proteins (15 weight%) 50% (without water) • Construction materials Collagen Spider silk proteins 3 Proteins Structural proteins structural role & mechanical support "cell skeleton“: complex network of protein filaments. muscle contraction results from action of large protein assemblies Myosin und Myogen. Other organic material (hair and bone) are also based on proteins. Collagen is found in all multi cellular animals, occurring in almost every tissue. • It is the most abundant vertebrate protein • approximately a quarter of mammalian protein is collagen 4 Proteins Storage Various ions, small molecules and other metabolites are stored by complexation with proteins • hemoglobin stores oxygen (free O2 in the blood would be toxic) • iron is stored by ferritin Transport Proteins are involved in the transportation of particles ranging from electrons to macromolecules. • Iron is transported by transferrin • Oxygen via hemoglobin. • Some proteins form pores in cellular membranes through which ions pass; the transport of proteins themselves across membranes also depends on other proteins. Beside: Regulation, enzymes, defense, functional properties 5 Our universal container system: Albumines 6 zones: IA, IB, IIA, IIB, IIIA, IIIB; IIB and -

Asian Journal of Chemistry Asian Journal of Chemistry
Asian Journal of Chemistry; Vol. 28, No. 1 (2016), 116-120 ASIAN JOURNAL OF CHEMISTRY http://dx.doi.org/10.14233/ajchem.2016.19262 Theoretical Studies on Interaction Between CO2 Gas and Imidazolium-Type Organic Ionic Liquid Using DFT and Natural Bond Orbital Calculations 1,2,* SAIED M. SOLIMAN 1Department of Chemistry, Rabigh College of Science and Art, King Abdulaziz University, Jeddah, P.O. Box 344, Rabigh 21911, Saudi Arabia 2Department of Chemistry, Faculty of Science, Alexandria University, P.O. Box 426 Ibrahimia, 21525 Alexandria, Egypt *Corresponding author: Tel: +966 565450752; E-mail: [email protected]; [email protected] Received: 20 April 2015; Accepted: 31 July 2015; Published online: 5 October 2015; AJC-17557 The interaction between 4,5-dibromo-1-butyl-3-methylimidazolium trifluoromethanesulfonate; [DBBIM][TFMSO3] ionic liquid and CO2 gas has been studied using DFT calculations. With the help of natural bond orbital (NBO) analyses, the second order perturbation energies of the most interacting natural bond orbitals and natural atomic charges have been predicted by the density functional theory (DFT) computations at the X3LYP/6-31++G(d,p) level of theory. The natural charge calculations showed that in the ionic liquid-CO2 system, the [DBBIM][TFMSO3] ionic liquid is the electron donor while CO2 gas is an electron acceptor. Further stabilization of the imidazolium ring π-system is produced due to the interaction of the CO2 gas with the ionic liquid. The polarization of the C–O and S1–O3 bonds are affected significantly by such interactions. The charge decomposition (CDA) analysis revealed the strong interactions between the [DBBIM]+ and – [TFMSO3] ions of the ionic liquid and the weak interaction between the ionic liquid and the CO2 gas. -

Resonance 1. Lone Pair Next to Empty 2P Orbital
1 Lecture 5 Resonance 1. Lone pair next to empty 2p orbital electron pair acceptors - lack of electrons C C CC sp2 R+ is more common sp R+ is less common R+ needs electrons, has to overlap with a. an adjacent 2p lone pair with electrons b. an adjacent pi bond a. an adjacent 2p lone pair with electrons on a neutral atom (+ overall, delocalization of positive charge) R R X = neutral atom with lone pair R R C C R X 2D resonance R X C C R X R X R R R R C R C R CX CX R N R N R 3D resonance R R R R R R R C R C R CX R O CX 3D resonance R O R R R R C better, more bonds, full octets C R F R F b. an adjacent 2p lone pair with electrons on a negative atom (neutral overall, delocalization of electrons) R R X =anion with lone pair R R C C R X 2D resonance R X C C R X R X R R R R C R C R CX CX R C R C R 3D resonance R R R R R R R C R C R CX R N CX 3D resonance R N R R R R C C better, more bonds, full octets R O R O 2 Lecture 5 Problem 1 – All of the following examples demonstrate delocalization of a lone pair of electrons into an empty 2p orbital. Usually in organic chemistry this is a carbocation site, but not always.