Shallow Valence Band of Rutile Geo2 and P-Type Doping
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

New Mineral Names
_._-- -- American Mineralogist, Volume 69, pages 406-412, 1984 NEW MINERAL NAMES. PETE J. DUNN, LOUIS J. CABRI, GEORGE Y. CHAO, MICHAEL FLEISCHER, CARL A. FRANCIS, JOEL D. GRICE, JOHN L. JAMBOR, AND ADOLF PABST Argutite. The name is for the locality, the Carboire deposit on the Carboirite. Marimana Dome. J.D.G. Z. Johan, E. Oudin and P. Picot (1983)Germanium and gallium anal()gues of silicates and oxides found in the zinc deposits of central Pyrenees, France; argutite and carboirite, two new Chessexite. ~ineral species. Tschermaks Min. Petrogr. Mitt., 31, 97-119 H. Sarp and J. Defern~ (1982) Chessexite, a new mineral. (in French). Schweiz. Mineral. Petrogr. Mitt., 62, 337-341 (in French). Argutite Analysis by atomic absorption (N a and K by flame photome- try, S by chromatography, and H20 by TGA) gave S03 31.91, The average of seven electron microprobe analyses yielded Si02 3.64, Ah03 15.30, MgO 3.50, ZnO 3.73, CaO 3.92, Na20 Ge02 95.82, Mn02 0.17, FeO 1.20, ZnO 3.03, sum 100.22. This 4.44, K20 0.30, H20 31.11, sum 97.85. This gives the formula gives the formula (GeO.942,Mno.OO2,FeO.018,ZIlo.038)1:101.944or (Na3.75, Ko.17, Cao.08) (Ca1.75, M80.2') (Mg1.87, Znt.20) (AI,.85, ideally Ge02. M80.1') (Si1.,8, SlO.42)1:120'3.46 45.13 H20 or ideally (Na,K)4 No X-ray studies were possible due to the small grain size. Ca2(Mg,Zn)3AI8(Si04)2(S04)1O(OH)10 · 40H20. -

CRITICAL METALS HANDBOOK Critical Metals Handbook
CRITICAL METALS HANDBOOK Critical Metals Handbook Edited by Gus Gunn British Geological Survey Keyworth Nottingham UK Published in collaboration with the British Geological Survey This work is a co-publication between the American Geophysical Union and Wiley This edition first published 2014 © 2014 by John Wiley & Sons, Ltd This work is a co-publication between the American Geophysical Union and Wiley Registered Office John Wiley & Sons, Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK Editorial Offices 9600 Garsington Road, Oxford, OX4 2DQ, UK The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK 111 River Street, Hoboken, NJ 07030-5774, USA For details of our global editorial offices, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com/wiley-blackwell. The right of the author to be identified as the author of this work has been asserted in accordance with the UK Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The publisher is not associated with any product or vendor mentioned in this book. -

Metallogenesis of Germanium—A Review ⁎ R
Ore Geology Reviews 30 (2007) 145–180 www.elsevier.com/locate/oregeorev Metallogenesis of germanium—A review ⁎ R. Höll a, , M. Kling b, E. Schroll c a Department für Geo-und Umweltwissenschaften, Sektion Geologie, LMU München, Luisenstrasse 37, D-80333 München, Germany b Department für Geo-und Umweltwissenschaften, Sektion Mineralogie, Petrologie und Geochemie, LMU München, Theresienstrasse 41, D-80333 München, Germany c Institut für Mineralogie und Kristallographie, Universität Wien, Althanstrasse 14, A-1090 Wien, Austria Received 5 November 2002; accepted 23 July 2005 Available online 15 June 2006 Abstract Germanium (Ge) is a scarce, but not an extremely rare element in the Earth's crust (about 1.6 ppm Ge crustal average). Principal geochemical substitutions and mineral associations of Ge include Si, C, Zn, Cu, Fe, Sn, and Ag. Most Ge is dispersed through silicate minerals due to the substitution of Ge4+ for the geochemically similar Si4+. Ge is unusual in that it exhibits siderophile, lithophile, chalcophile and organophile behaviour in different geologic environments. Only minor variations in Ge concentrations are known from different igneous rocks, siliceous sedimentary rocks, and their metamorphic equivalents. Carbonates and evaporites show a depletion to the crustal average of Ge. There is a tendency for Ge to be slightly enriched in silicate minerals of late magmatic differentiates (e.g., muscovite granites), rocks that crystallize in the presence of a high volatile concentration (e.g., pegmatites, greisens) and late hydrothermal -

IMA–CNMNC Approved Mineral Symbols
Mineralogical Magazine (2021), 85, 291–320 doi:10.1180/mgm.2021.43 Article IMA–CNMNC approved mineral symbols Laurence N. Warr* Institute of Geography and Geology, University of Greifswald, 17487 Greifswald, Germany Abstract Several text symbol lists for common rock-forming minerals have been published over the last 40 years, but no internationally agreed standard has yet been established. This contribution presents the first International Mineralogical Association (IMA) Commission on New Minerals, Nomenclature and Classification (CNMNC) approved collection of 5744 mineral name abbreviations by combining four methods of nomenclature based on the Kretz symbol approach. The collection incorporates 991 previously defined abbreviations for mineral groups and species and presents a further 4753 new symbols that cover all currently listed IMA minerals. Adopting IMA– CNMNC approved symbols is considered a necessary step in standardising abbreviations by employing a system compatible with that used for symbolising the chemical elements. Keywords: nomenclature, mineral names, symbols, abbreviations, groups, species, elements, IMA, CNMNC (Received 28 November 2020; accepted 14 May 2021; Accepted Manuscript published online: 18 May 2021; Associate Editor: Anthony R Kampf) Introduction used collection proposed by Whitney and Evans (2010). Despite the availability of recommended abbreviations for the commonly Using text symbols for abbreviating the scientific names of the studied mineral species, to date < 18% of mineral names recog- chemical elements -
New Data on Minerals
Russian Academy of Science Fersman Mineralogical Museum Volume 39 New Data on Minerals Founded in 1907 Moscow Ocean Pictures Ltd. 2004 ISBN 5900395626 UDC 549 New Data on Minerals. Moscow.: Ocean Pictures, 2004. volume 39, 172 pages, 92 color images. EditorinChief Margarita I. Novgorodova. Publication of Fersman Mineralogical Museum, Russian Academy of Science. Articles of the volume give a new data on komarovite series minerals, jarandolite, kalsilite from Khibiny massif, pres- ents a description of a new occurrence of nikelalumite, followed by articles on gemnetic mineralogy of lamprophyl- lite barytolamprophyllite series minerals from IujaVritemalignite complex of burbankite group and mineral com- position of raremetaluranium, berrillium with emerald deposits in Kuu granite massif of Central Kazakhstan. Another group of article dwells on crystal chemistry and chemical properties of minerals: stacking disorder of zinc sulfide crystals from Black Smoker chimneys, silver forms in galena from Dalnegorsk, tetragonal Cu21S in recent hydrothermal ores of MidAtlantic Ridge, ontogeny of spiralsplit pyrite crystals from Kursk magnetic Anomaly. Museum collection section of the volume consist of articles devoted to Faberge lapidary and nephrite caved sculp- tures from Fersman Mineralogical Museum. The volume is of interest for mineralogists, geochemists, geologists, and to museum curators, collectors and ama- teurs of minerals. EditorinChief Margarita I .Novgorodova, Doctor in Science, Professor EditorinChief of the volume: Elena A.Borisova, Ph.D Editorial Board Moisei D. Dorfman, Doctor in Science Svetlana N. Nenasheva, Ph.D Marianna B. Chistyakova, Ph.D Elena N. Matvienko, Ph.D Мichael Е. Generalov, Ph.D N.A.Sokolova — Secretary Translators: Dmitrii Belakovskii, Yiulia Belovistkaya, Il'ya Kubancev, Victor Zubarev Photo: Michael B. -

Indications of the Magnetic State in the Charge Distributions in Mno, Coo, and Nio. I: Para- and Antiferromagnetism of Mno1
Crystallography Reports, Vol. 47, No. 3, 2002, pp. 347–361. From Kristallografiya, Vol. 47, No. 3, 2002, pp. 391–405. Original English Text Copyright © 2002 by Jean-Pierre Vidal, Genevieve Vidal-Valat, Kaarle Kurki-Suonio, Riitta Kurki-Suonio. DIFFRACTION AND SCATTERING OF X-RAY AND SYNCHROTRON RADIATION To the Memory of Riitta Kurki-Suonio Indications of the Magnetic State in the Charge Distributions in MnO, CoO, and NiO. I: Para- and Antiferromagnetism of MnO1 Jean-Pierre Vidal*, Genevieve Vidal-Valat*, Kaarle Kurki-Suonio**, and Riitta Kurki-Suonio**† * Laboratoire d’Analyse Multipolaire des Repartitions de Charges Experimentales, Université Montpellier 2, 34095 Montpellier Cedex 05, France e-mail: [email protected] ** Physics Department, P.O. Box 9, FIN-00014 University of Helsinki, Finland Received May 28, 2001 Abstract—X-ray diffraction intensities from MnO and CoO were measured above and below their Néel tem- peratures and from NiO, below the Néel temperature. To detect possible characteristics of the paramagnetic and antiferromagnetic states of the crystals, the data were subjected to direct multipole analysis of the atomic- charge densities. For MnO, both spherical and nonspherical accumulation-of-charge densities indicate the exchange of the roles played by manganese and oxygen in the magnetic phase transition. Both spherical and nonspherical features characteristic of the ionic nature are inherent in both states. The electron counts of the density peaks correspond to Mn2+ and O1–, with the tenth electron of O2– being distributed in a wider region. In the paramagnetic state, there is an electronic Mn–Mn bond which seems to be formed due to coupling with the tenth electron of O2– and builds up a three-dimensional net of the charge density with the “cages” surround- ing oxygen atoms. -

List of Mineral Symbols
THE CANADIAN MINERALOGIST LIST OF SYMBOLS FOR ROCK- AND ORE-FORMING MINERALS (January 1, 2021) ____________________________________________________________________________________________________________ Ac acanthite Ado andorite Asp aspidolite Btr berthierite Act actinolite Adr andradite Ast astrophyllite Brl beryl Ae aegirine Ang angelaite At atokite Bll beryllonite AeAu aegirine-augite Agl anglesite Au gold Brz berzelianite Aen aenigmatite Anh anhydrite Aul augelite Bet betafite Aes aeschynite-(Y) Ani anilite Aug augite Bkh betekhtinite Aik aikinite Ank ankerite Aur auricupride Bdt beudantite Akg akaganeite Ann annite Aus aurostibite Beu beusite Ak åkermanite An anorthite Aut autunite Bch bicchulite Ala alabandite Anr anorthoclase Aw awaruite Bt biotite* Ab albite Atg antigorite Axn axinite-(Mn) Bsm bismite Alg algodonite Sb antimony Azu azurite Bi bismuth All allactite Ath anthophyllite Bdl baddeleyite Bmt bismuthinite Aln allanite Ap apatite* Bns banalsite Bod bohdanowiczite Alo alloclasite Arg aragonite Bbs barbosalite Bhm böhmite Ald alluaudite Ara aramayoite Brr barrerite Bor boralsilite Alm almandine Arf arfvedsonite Brs barroisite Bn bornite Alr almarudite Ard argentodufrénoysite Blt barylite Bou boulangerite Als alstonite Apn argentopentlandite Bsl barysilite Bnn bournonite Alt altaite Arp argentopyrite Brt baryte, barite Bow bowieite Aln alunite Agt argutite Bcl barytocalcite Brg braggite Alu alunogen Agy argyrodite Bss bassanite Brn brannerite Amb amblygonite Arm armangite Bsn bastnäsite Bra brannockite Ams amesite As arsenic -

Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro
minerals Article Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro-XRF Mapping and Micro-XANES Spectroscopy Rémi Belissont 1,2, Manuel Munoz 3 , Marie-Christine Boiron 1,*, Béatrice Luais 2 and Olivier Mathon 4 1 Université de Lorraine, CNRS, GeoRessources, F-54000 Nancy, France; [email protected] 2 Centre de Recherches Pétrographiques et Géochimiques (CRPG), Université de Lorraine, CNRS, UMR 7358, 15 Rue Notre Dame des Pauvres, BP 20, CEDEX, F-54501 Vandoeuvre-lès-Nancy, France; [email protected] 3 Geosciences Montpellier, Université de Montpellier, CNRS, CEDEX 5, 34095 Montpellier, France; [email protected] 4 European Synchrotron Radiation Facility, BP 220, F-38043 Grenoble, France; [email protected] * Correspondence: [email protected] Received: 18 March 2019; Accepted: 9 April 2019; Published: 12 April 2019 Abstract: Germanium is considered a critical element, with a demand that has sharply increased due to booming high-technology industries. To understand Ge incorporation mechanisms in natural systems, we investigate Ge speciation in Cu-bearing sulfide minerals using synchrotron X-ray fluorescence (XRF) chemical mapping and Ge K-edge µ-X-ray absorption near-edge structures (µ-XANES) spectroscopy. The samples investigated include (i) a homogeneous chalcopyrite from the Kipushi polymetallic deposit (Central African copperbelt, D.R. Congo) and (ii) a zoned Ge-rich chalcopyrite from the Barrigão Cu deposit (Iberian pyrite belt, Portugal). First, our spectroscopic analysis supports the occurrence of tetrahedrally-coordinated Ge4+ in chalcopyrite, independently from origins or zoning patterns observed for these minerals. Then, based on statistical analyses of XRF chemical maps, we demonstrate that tetravalent germanium most likely incorporates chalcopyrite through the Fe crystallographic site via coupled substitutions with the following form: (2x + 3y)Fe3+ $ (x + 2y)(Ge,Sn)4+ + x(Zn,Pb)2+ + y(Cu,Ag)+, although the presence of lattice vacancies cannot be completely excluded. -

Vibrational Dynamics of Rutile-Type Geo2 from Micro-Raman Spectroscopy Experiments and First-Principles Calculations
Home Search Collections Journals About Contact us My IOPscience Vibrational dynamics of rutile-type GeO2 from micro-Raman spectroscopy experiments and first-principles calculations This content has been downloaded from IOPscience. Please scroll down to see the full text. 2015 EPL 109 26007 (http://iopscience.iop.org/0295-5075/109/2/26007) View the table of contents for this issue, or go to the journal homepage for more Download details: IP Address: 157.27.226.238 This content was downloaded on 02/04/2015 at 07:57 Please note that terms and conditions apply. January 2015 EPL, 109 (2015) 26007 www.epljournal.org doi: 10.1209/0295-5075/109/26007 Vibrational dynamics of rutile-type GeO2 from micro-Raman spectroscopy experiments and first-principles calculations A. Sanson1,G.S.Pokrovski2,M.Giarola3 and G. Mariotto3 1 Department of Physics and Astronomy, University of Padova - Via Marzolo 8, 35131 Padova, Italy 2 Groupe M´etallog´enie Exp´erimentale, G´eosciences Environnement Toulouse GET, University of Toulouse, CNRS - IRD - OMP - Avenue Edouard Belin 14, 31400 Toulouse, France 3 Department of Computer Science, University of Verona - Strada le Grazie 15, 37134 Verona, Italy received 4 December 2014; accepted in final form 18 January 2015 published online 3 February 2015 PACS 63.70.+h – Statistical mechanics of lattice vibrations and displacive phase transitions PACS 78.30.Er – Infrared and Raman spectra: Solid metals and alloys PACS 78.20.-e – Optical properties of bulk materials and thin films Abstract – The vibrational dynamics of germanium dioxide in the rutile structure has been investigated by using polarized micro-Raman scattering spectroscopy coupled with first-principles calculations. -

AND ORE-FORMING MINERALS (December 30, 2019) ______
THE CANADIAN MINERALOGIST LIST OF SYMBOLS FOR ROCK- AND ORE-FORMING MINERALS (December 30, 2019) ____________________________________________________________________________________________________________ Ac acanthite Adr andradite Ast astrophyllite Brl beryl Act actinolite Ang angelaite At atokite Bll beryllonite Ae aegirine Agl anglesite Au gold Brz berzelianite Aen aenigmatite Anh anhydrite Aul augelite Bet betafite Aes aeschynite-(Y) Ani anilite Aug augite Bkh betekhtinite Aik aikinite Ank ankerite Aur auricupride Bdt beudantite Akg akaganeite Ann annite Aus aurostibite Beu beusite Ak åkermanite An anorthite Aut autunite Bch bicchulite Ala alabandite Anr anorthoclase Aw awaruite Bt biotite* Ab albite Atg antigorite Axn axinite-(Mn) Bsm bismite Alg algodonite Sb antimony Azu azurite Bi bismuth All allactite Ath anthophyllite Bdl baddeleyite Bmt bismuthinite Aln allanite Ap apatite* Bns banalsite Bod bohdanowiczite Alo alloclasite Arg aragonite Bbs barbosalite Bhm böhmite Ald alluaudite Ara aramayoite Brr barrerite Bor boralsilite Alm almandine Arf arfvedsonite Brs barroisite Bn bornite Alr almarudite Ard argentodufrénoysite Blt barylite Bou boulangerite Als alstonite Apn argentopentlandite Bsl barysilite Bnn bournonite Alt altaite Arp argentopyrite Brt baryte, barite Bow bowieite Aln alunite Agt argutite Bcl barytocalcite Brg braggite Alu alunogen Agy argyrodite Bss bassanite Brn brannerite Amb amblygonite Arm armangite Bsn bastnäsite Bra brannockite Ams amesite As arsenic Bau baumstarkite Bnt braunite Amp amphibole* Ass arseniosiderite -
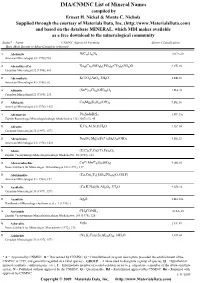
IMA/CNMNC List of Mineral Names
IMA/CNMNC List of Mineral Name s compiled by Ernest H. Nickel & Monte C. Nichols Supplied through the courtesy of Materials Data, Inc. (http://www.MaterialsData.com) and based on the database MINERAL, which MDI makes available as a free download to the mineralogical community Status* Name CNMNC Approved Formula Strunz Classification Best, Most Recent or Most Complete reference. A Abelsonite NiC£¡H£¢N¤ 10.CA.20 American Mineralogist 63 (1978) 930 A Abenakiite-(Ce) Na¢¦Ce¦(SiO£)¦(PO¤)¦(CO£)¦(SO¢)O 9.CK.10 Canadian Mineralogist 32 (1994), 843 G Abernathyite K(UO¢)AsO¤•3H¢O 8.EB.15 American Mineralogist 41 (1956), 82 A Abhurite (SnÀÈ)¢¡Cl¡¦(OH)¡¤O¦ 3.DA.30 Canadian Mineralogist 23 (1985), 233 D Abkhazite Ca¢Mg¥Si¨O¢¢(OH)¢ 9.DE.10 American Mineralogist 63 (1978), 1023 A Abramovite Pb¢SnInBiS§ 2.HF.25a Zapiski Rossiiskogo Mineralogicheskogo Obshchetstva 136 (2007) (5), 45 D Abrazite K,Ca,Al,Si,O,H¢O 9.GC.05 Canadian Mineralogist 35 (1997), 1571 D Abriachanite Na¢(Fe,Mg)£(FeÁÈ)¢Si¨O¢¢(OH)¢ 9.DE.25 American Mineralogist 63 (1978), 1023 D Absite (U,Ca,Y,Ce)(Ti,Fe)¢O¦ Zapiski Vsesoyuznogo Mineralogicheskogo Obshchestva 92 (1963), 113 A Abswurmbachite CuÀÈ(MnÁÈ)¦O¨(SiO¤) 9.AG.05 Neues Jahrbuch für Mineralogie, Abhandlungen 163 (1991), 117 D Abukumalite (Ca,Ce)¢Y£(SiO¤,PO¤)£(O,OH,F) American Mineralogist 51 (1966), 152 D Acadialite (Ca,K,Na)(Si,Al)£O¦•3H¢O 9.GD.10 Canadian Mineralogist 35 (1997), 1571 G Acanthite Ag¢S 2.BA.30a Handbook of Mineralogy (Anthony et al.), 1 (1990), 1 A Acetamide CH£CONH¢ 10.AA.20 Zapiski Vsesoyuznogo Mineralogicheskogo -

Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro
minerals Article Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro-XRF Mapping and Micro-XANES Spectroscopy Rémi Belissont 1,2, Manuel Munoz 3 , Marie-Christine Boiron 1,*, Béatrice Luais 2 and Olivier Mathon 4 1 Université de Lorraine, CNRS, GeoRessources, F-54000 Nancy, France; [email protected] 2 Centre de Recherches Pétrographiques et Géochimiques (CRPG), Université de Lorraine, CNRS, UMR 7358, 15 Rue Notre Dame des Pauvres, BP 20, CEDEX, F-54501 Vandoeuvre-lès-Nancy, France; [email protected] 3 Geosciences Montpellier, Université de Montpellier, CNRS, CEDEX 5, 34095 Montpellier, France; [email protected] 4 European Synchrotron Radiation Facility, BP 220, F-38043 Grenoble, France; [email protected] * Correspondence: [email protected] Received: 18 March 2019; Accepted: 9 April 2019; Published: 12 April 2019 Abstract: Germanium is considered a critical element, with a demand that has sharply increased due to booming high-technology industries. To understand Ge incorporation mechanisms in natural systems, we investigate Ge speciation in Cu-bearing sulfide minerals using synchrotron X-ray fluorescence (XRF) chemical mapping and Ge K-edge µ-X-ray absorption near-edge structures (µ-XANES) spectroscopy. The samples investigated include (i) a homogeneous chalcopyrite from the Kipushi polymetallic deposit (Central African copperbelt, D.R. Congo) and (ii) a zoned Ge-rich chalcopyrite from the Barrigão Cu deposit (Iberian pyrite belt, Portugal). First, our spectroscopic analysis supports the occurrence of tetrahedrally-coordinated Ge4+ in chalcopyrite, independently from origins or zoning patterns observed for these minerals. Then, based on statistical analyses of XRF chemical maps, we demonstrate that tetravalent germanium most likely incorporates chalcopyrite through the Fe crystallographic site via coupled substitutions with the following form: (2x + 3y)Fe3+ $ (x + 2y)(Ge,Sn)4+ + x(Zn,Pb)2+ + y(Cu,Ag)+, although the presence of lattice vacancies cannot be completely excluded.