SROC Chapter 2
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

1,1,1,2-Tetrafluoroethane
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Concise International Chemical Assessment Document 11 1,1,1,2-Tetrafluoroethane First draft prepared by Mrs P. Barker and Mr R. Cary, Health and Safety Executive, Liverpool, United Kingdom, and Dr S. Dobson, Institute of Terrestrial Ecology, Huntingdon, United Kingdom Please not that the layout and pagination of this pdf file are not identical to the printed CICAD Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 1998 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organisation (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization -

SAFETY DATA SHEET Difluoromethane (R32) SECTION 1
SAFETY DATA SHEET Difluoromethane (R32) Issue Date: 16.01.2013 Version: 1.1 SDS No.: 000010021734 Last revised date: 26.11.2018 1/14 SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifier Product name: Difluoromethane (R32) Other Name: HFC-32 Additional identification Chemical name: Difluoromethane Chemical formula: CH2F2 INDEX No. - CAS-No. 75-10-5 EC No. 200-839-4 REACH Registration No. 01-2119471312-47 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses: Industrial and professional. Perform risk assessment prior to use. Refrigerant. Use as an Intermediate (transported, on-site isolated). Use for electronic component manufacture. Using gas alone or in mixtures for the calibration of analysis equipment. Formulation of mixtures with gas in pressure receptacles. Uses advised against Consumer use. 1.3 Details of the supplier of the safety data sheet Supplier Linde Gas GmbH Telephone: +43 50 4273 Carl-von-Linde-Platz 1 A-4651 Stadl-Paura E-mail: [email protected] 1.4 Emergency telephone number: Emergency number Linde: + 43 50 4273 (during business hours), Poisoning Information Center: +43 1 406 43 43 SECTION 2: Hazards identification 2.1 Classification of the substance or mixture Classification according to Regulation (EC) No 1272/2008 as amended. Physical Hazards Flammable gas Category 1 H220: Extremely flammable gas. Gases under pressure Liquefied gas H280: Contains gas under pressure; may explode if heated. SDS_AT - 000010021734 SAFETY DATA SHEET Difluoromethane (R32) Issue Date: 16.01.2013 Version: 1.1 SDS No.: 000010021734 Last revised date: 26.11.2018 2/14 2.2 Label Elements Signal Words: Danger Hazard Statement(s): H220: Extremely flammable gas. -

Chlorofluorocarbons Chem 300A Tyler Jensen, Mackenzie Latimer, Jessie Luther, Jake Mcghee, Adeeb Noorani, Tyla Penner, & Maddy Springle
Chlorofluorocarbons Chem 300a Tyler Jensen, Mackenzie Latimer, Jessie Luther, Jake McGhee, Adeeb Noorani, Tyla Penner, & Maddy Springle 1.1: A Brief History of Chlorofluorocarbons Chlorofluorocarbons (CFCs), also known as Freons, were first synthesized in 1928 by Thomas Midgley Jr, who was working for General Motors trying to find a safe refrigerant to use in commercial applications. (Rosenbaum, n.d.). They are an anthropogenic compound containing fluorine, carbon, and chlorine atoms, and are classified as halocarbons. CFCs are a family of chemicals based upon hydrocarbon skeletons, where most hydrogens have been replaced with chlorine and/or fluorine atoms. They are chemically stable freons that are non-flammable, tasteless and odourless. CFCs are very volatile, which makes for ideal refrigerant gases, having a boiling point close to zero degrees (Rosenbaum, n.d) They were originally created to replace the toxic gases used in the late 1800’s and early 1900’s. Examples of the toxic gases replaced by CFCs as refrigerants are ammonia (NH3), methyl chloride (CH3Cl), and sulfur dioxide (SO2) (Wilkins, 1999). When first created dichlorodifloromethane was found to be less toxic than carbon dioxide, and as non-flammable as carbon tetrachloride (Midgley & Henne, 1930). The non-toxic, non-flammable, and non-reactive properties of CFCs made them ideal for use as refrigerants. CFCs were used in many developed countries for consumption and production as they were inflammable and non-toxic towards humanity. Chlorofluorocarbons can be used as refrigerants, cleaning agents, foaming agents, and propellants for aerosol sprays (Welch, n.d.). In 1974, Two University of California chemists, Professor F. Sherwood Rowland and Dr. -

AN ACT Relating to Reducing Greenhouse Gas Emissions from 1
H-0017.2 HOUSE BILL 1050 State of Washington 67th Legislature 2021 Regular Session By Representatives Fitzgibbon, Ortiz-Self, Leavitt, Duerr, Chopp, Ramel, Peterson, Goodman, Ryu, Callan, Ramos, Ormsby, Pollet, Stonier, Fey, Macri, and Bergquist Prefiled 12/23/20. Read first time 01/11/21. Referred to Committee on Environment & Energy. 1 AN ACT Relating to reducing greenhouse gas emissions from 2 fluorinated gases; amending RCW 70A.15.6410, 70A.15.6420, 3 70A.15.6430, 70A.45.080, 19.27.580, 70A.15.3150, 70A.15.3160, 4 19.285.040, 19.27A.220, and 39.26.310; reenacting and amending RCW 5 70A.45.010; adding a new chapter to Title 70A RCW; creating a new 6 section; and recodifying RCW 70A.45.080, 70A.15.6410, 70A.15.6420, and 70A.15.6430.7 8 BE IT ENACTED BY THE LEGISLATURE OF THE STATE OF WASHINGTON: 9 NEW SECTION. Sec. 1. (1) The legislature finds that 10 hydrofluorocarbons are air pollutants that pose significant threats 11 to our environment. Although hydrofluorocarbons currently represent a 12 small proportion of the state's greenhouse gas emissions, emissions 13 of hydrofluorocarbons have been rapidly increasing in the United 14 States and worldwide, and they are hundreds to thousands of times 15 more potent than carbon dioxide. In 2019, the legislature took a 16 significant step towards reducing greenhouse gas emissions from 17 hydrofluorocarbons by transitioning to the use of less damaging 18 hydrofluorocarbons or suitable substitutes in certain new foam, 19 aerosol, and refrigerant uses. However, significant sources of 20 hydrofluorocarbon emissions in Washington remain unaddressed by the 21 2019 legislation, including legacy uses of hydrofluorocarbons as a p. -

JSP 418 Leaflet 6 Fluorinated Greenhouse Gases Version
Management of Environmental Protection in Leaflet 6 JSP 418 Defence FLUORINATED GREENHOUSE GASES Contents Para 1 Introduction Policies 12 International Policy – The UN Framework Convention on Climate Change 15 European Union Policy 17 UK Government Policy 23 MOD Policy Legislation 25 International legislation 45 UK Legislation Procedures for Implementation of MOD Policy 49 Policy Development and Implementation Management Responsibilities 51 Responsibilities of Users 52 Responsibilities of MOD Re-Supplier (reselling to MOD purchasers) 55 Training 73 Restrictions on New Uses 77 Restrictions on Existing Uses 79 Alternative Substances and Methods 80 Responsible Management 83 Servicing Requirements 86 Reporting Requirements 89 Disposal of Recovered and Unwanted Fluorinated Greenhouse Gases 91 Further Guidance Annexes A Definitions from Regulation (EU) No 517/2014 B Placing on the market prohibitions in accordance with Article 9 of Regulation EC842/2006 C Common and Trade Names of Products That May Contain Fluorinated Greenhouse Gases D Assurance Questions May 2016 Leaflet 6 Page 1 Management of Environmental Protection in Leaflet 6 JSP 418 Defence INTRODUCTION Aim 1. The aim of this leaflet is to deliver MOD policy requirements on the use, containment and recovery of fluorinated greenhouse gases (F gases). It also outlines the latest legislative position and the substances whose use and applications are now prohibited. Scope 2. This leaflet applies to all personnel including Project Team Leaders, Project Sponsors, Equipment, Property and Facilities Managers and others (including Regional Prime Contractors RPCs, Private Partners and other such contractors who operate and use equipment containing fluorinated greenhouse gases. This includes those who take the role of undertakings1 and/or a Responsible Authority2 who manages such undertakings and operate equipment or facilities. -

Avoiding Fluorinated Greenhouse Gases Prospects for Phasing Out
| CLIMATE CHANGE | 08/2011 Avoiding Fluorinated Greenhouse Gases Prospects for Phasing Out | CLIMATE CHANGE | 08/2011 Avoiding Fluorinated Greenhouse Gases Prospects for Phasing Out by Katja Becken Dr. Daniel de Graaf Dr. Cornelia Elsner Gabriele Hoffmann Dr. Franziska Krüger Kerstin Martens Dr. Wolfgang Plehn Dr. Rolf Sartorius German Federal Environment Agency (Umweltbundesamt) UMWELTBUNDESAMT This publication is only available online. It can be downloaded from http://www.uba.de/uba-info-medien-e/3977.html along with a German version. Revised version of the report “Fluorinated Greenhouse Gases in Products and Processes – Technical Climate Protection Measures”, German Federal Environment Agency, Berlin 2004 Translation of the German-language report, November 2010 ISSN 1862-4359 Publisher: Federal Environment Agency (Umweltbundesamt) Wörlitzer Platz 1 06844 Dessau-Roßlau Germany Phone: +49-340-2103-0 Fax: +49-340-2103 2285 Email: [email protected] Internet: http://www.umweltbundesamt.de http://fuer-mensch-und-umwelt.de/ Edited by: Section III 1.4 Substance-related Product Issues Katja Becken, Dr. Wolfgang Plehn Dessau-Roßlau, June 2011 Foreword Fluorinated greenhouse gases (F-gases) are 100 to 24,000 times more harmful to the climate than CO2. The contribution of fluorinated greenhouse gases to global warming is projected to triple from nearly 2% to around 6% of total greenhouse gas emissions by the year 2050. This is revealed by global projections prepared for the Federal Environment Agency in a scenario where no new measures are taken. The need for action is evident. F-gases are mostly used in similar ways to the CFCs and halons used in the past, which are responsible for the destruction of the ozone layer in the stratosphere. -

Secondary Organic Aerosol from Chlorine-Initiated Oxidation of Isoprene Dongyu S
Secondary organic aerosol from chlorine-initiated oxidation of isoprene Dongyu S. Wang and Lea Hildebrandt Ruiz McKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, TX 78756, USA 5 Correspondence to: Lea Hildebrandt Ruiz ([email protected]) Abstract. Recent studies have found concentrations of reactive chlorine species to be higher than expected, suggesting that atmospheric chlorine chemistry is more extensive than previously thought. Chlorine radicals can interact with HOx radicals and nitrogen oxides (NOx) to alter the oxidative capacity of the atmosphere. They are known to rapidly oxidize a wide range of volatile organic compounds (VOC) found in the atmosphere, yet little is known about secondary organic aerosol (SOA) 10 formation from chlorine-initiated photo-oxidation and its atmospheric implications. Environmental chamber experiments were carried out under low-NOx conditions with isoprene and chlorine as primary VOC and oxidant sources. Upon complete isoprene consumption, observed SOA yields ranged from 8 to 36 %, decreasing with extended photo-oxidation and SOA aging. Formation of particulate organochloride was observed. A High-Resolution Time-of-Flight Chemical Ionization Mass Spectrometer was used to determine the molecular composition of gas-phase species using iodide-water and hydronium-water 15 cluster ionization. Multi-generational chemistry was observed, including ions consistent with hydroperoxides, chloroalkyl hydroperoxides, isoprene-derived epoxydiol (IEPOX) and hypochlorous acid (HOCl), evident of secondary OH production and resulting chemistry from Cl-initiated reactions. This is the first reported study of SOA formation from chlorine-initiated oxidation of isoprene. Results suggest that tropospheric chlorine chemistry could contribute significantly to organic aerosol loading. -

Purification Process of Pentafluoroethane (HFC-125) Verfahren Zur Reinigung Von Pentafluorethan (HFC-125) Procédé De Purification Du Pentafluoroéthane (HFC-125)
Europäisches Patentamt *EP001153907B1* (19) European Patent Office Office européen des brevets (11) EP 1 153 907 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.7: C07C 17/386 of the grant of the patent: 06.10.2004 Bulletin 2004/41 (21) Application number: 01109907.4 (22) Date of filing: 24.04.2001 (54) Purification process of pentafluoroethane (HFC-125) Verfahren zur Reinigung von Pentafluorethan (HFC-125) Procédé de purification du pentafluoroéthane (HFC-125) (84) Designated Contracting States: • Basile, Giampiero DE ES FR GB IT NL 15100 Alessandria (IT) (30) Priority: 09.05.2000 IT MI001006 (74) Representative: Jacques, Philippe et al Solvay S.A. (43) Date of publication of application: Département de la Propriété Industrielle, 14.11.2001 Bulletin 2001/46 Rue de Ransbeek, 310 1120 Bruxelles (BE) (73) Proprietor: Solvay Solexis S.p.A. 20121 Milano (IT) (56) References cited: EP-A- 0 626 362 EP-A- 0 985 650 (72) Inventors: • Azzali, Daniele 20021 Bollate (MI) (IT) Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. Notice of opposition shall be filed in a written reasoned statement. It shall not be deemed to have been filed until the opposition fee has been paid. (Art. 99(1) European Patent Convention). EP 1 153 907 B1 Printed by Jouve, 75001 PARIS (FR) EP 1 153 907 B1 Description [0001] The present invention relates to a purification process of pentafluoroethane (HFC-125) containing as impurity chloropentafluoroethane (CFC-115). -

Use of Chlorofluorocarbons in Hydrology : a Guidebook
USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A Guidebook USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A GUIDEBOOK 2005 Edition The following States are Members of the International Atomic Energy Agency: AFGHANISTAN GREECE PANAMA ALBANIA GUATEMALA PARAGUAY ALGERIA HAITI PERU ANGOLA HOLY SEE PHILIPPINES ARGENTINA HONDURAS POLAND ARMENIA HUNGARY PORTUGAL AUSTRALIA ICELAND QATAR AUSTRIA INDIA REPUBLIC OF MOLDOVA AZERBAIJAN INDONESIA ROMANIA BANGLADESH IRAN, ISLAMIC REPUBLIC OF RUSSIAN FEDERATION BELARUS IRAQ SAUDI ARABIA BELGIUM IRELAND SENEGAL BENIN ISRAEL SERBIA AND MONTENEGRO BOLIVIA ITALY SEYCHELLES BOSNIA AND HERZEGOVINA JAMAICA SIERRA LEONE BOTSWANA JAPAN BRAZIL JORDAN SINGAPORE BULGARIA KAZAKHSTAN SLOVAKIA BURKINA FASO KENYA SLOVENIA CAMEROON KOREA, REPUBLIC OF SOUTH AFRICA CANADA KUWAIT SPAIN CENTRAL AFRICAN KYRGYZSTAN SRI LANKA REPUBLIC LATVIA SUDAN CHAD LEBANON SWEDEN CHILE LIBERIA SWITZERLAND CHINA LIBYAN ARAB JAMAHIRIYA SYRIAN ARAB REPUBLIC COLOMBIA LIECHTENSTEIN TAJIKISTAN COSTA RICA LITHUANIA THAILAND CÔTE D’IVOIRE LUXEMBOURG THE FORMER YUGOSLAV CROATIA MADAGASCAR REPUBLIC OF MACEDONIA CUBA MALAYSIA TUNISIA CYPRUS MALI TURKEY CZECH REPUBLIC MALTA UGANDA DEMOCRATIC REPUBLIC MARSHALL ISLANDS UKRAINE OF THE CONGO MAURITANIA UNITED ARAB EMIRATES DENMARK MAURITIUS UNITED KINGDOM OF DOMINICAN REPUBLIC MEXICO GREAT BRITAIN AND ECUADOR MONACO NORTHERN IRELAND EGYPT MONGOLIA UNITED REPUBLIC EL SALVADOR MOROCCO ERITREA MYANMAR OF TANZANIA ESTONIA NAMIBIA UNITED STATES OF AMERICA ETHIOPIA NETHERLANDS URUGUAY FINLAND NEW ZEALAND UZBEKISTAN FRANCE NICARAGUA VENEZUELA GABON NIGER VIETNAM GEORGIA NIGERIA YEMEN GERMANY NORWAY ZAMBIA GHANA PAKISTAN ZIMBABWE The Agency’s Statute was approved on 23 October 1956 by the Conference on the Statute of the IAEA held at United Nations Headquarters, New York; it entered into force on 29 July 1957. The Headquarters of the Agency are situated in Vienna. -

CHAPTER12 Atmospheric Degradation of Halocarbon Substitutes
CHAPTER12 AtmosphericDegradation of Halocarbon Substitutes Lead Author: R.A. Cox Co-authors: R. Atkinson G.K. Moortgat A.R. Ravishankara H.W. Sidebottom Contributors: G.D. Hayman C. Howard M. Kanakidou S .A. Penkett J. Rodriguez S. Solomon 0. Wild CHAPTER 12 ATMOSPHERIC DEGRADATION OF HALOCARBON SUBSTITUTES Contents SCIENTIFIC SUMMARY ......................................................................................................................................... 12.1 12.1 BACKGROUND ............................................................................................................................................... 12.3 12.2 ATMOSPHERIC LIFETIMES OF HFCS AND HCFCS ................................................................................. 12.3 12.2. 1 Tropospheric Loss Processes .............................................................................................................. 12.3 12.2.2 Stratospheric Loss Processes .............................................................................................................. 12.4 12.3 ATMOSPHERIC LIFETIMES OF OTHER CFC AND HALON SUBSTITUTES ......................................... 12.4 12.4 ATMOSPHERIC DEGRADATION OF SUBSTITUTES ................................................................................ 12.5 12.5 GAS PHASE DEGRADATION CHEMISTRY OF SUBSTITUTES .............................................................. 12.6 12.5.1 Reaction with NO .............................................................................................................................. -

Hfcs for THERMAL INSULATION
HFCs FOR THERMAL INSULATION Visit our web site www.fluorocarbons.org A SOLUTION ADDRESSING THE CLIMATE CHANGE CHALLENGE EFCTC - European FluoroCarbon Technical Committee Avenue E.van Nieuwenhuyse 4 B-1160 Brussels - Phone : +32 2 676 72 11 HFCs FOR THERMAL INSULATION Visit our web site www.fluorocarbons.org A SOLUTION ADDRESSING THE CLIMATE CHANGE CHALLENGE EFCTC - European FluoroCarbon Technical Committee Avenue E.van Nieuwenhuyse 4 B-1160 Brussels - Phone : +32 2 676 72 11 Progress for our quality of life… Could we do without heating or air conditioning? …and climate protection Refrigeration, air conditioning and heating are often essential to life particularly in public areas such as The greenhouse effect to a great extent determines the climate in hospitals and laboratories, for food products on earth. Growth in emissions of greenhouse gases associated with in the cold chain, for medical and computer equipment. human activities threa tens the climate balance. Carbon dioxide (CO2) - the main greenhouse gas - is emitted when fossil fuels are burnt to But we have also to consider the impact of produce energy and increasing energy demands have led to rapid growth the wide-spread use of these services on in the amount of CO2 in the atmosphere. Heating, air conditioning the global environment. and refrigeration have contributed to this growth. Roof If no action is taken at all, greenhouse gas(*) emissions could be expected to further increase in EU Member States by 17% 22% between 1990 and 2010, while the target set by the Kyoto Protocol for the period is to reduce the emissions by 8%. -
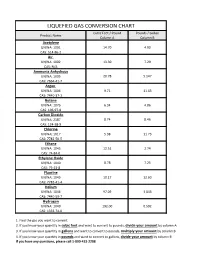
Liquefied Gas Conversion Chart
LIQUEFIED GAS CONVERSION CHART Cubic Feet / Pound Pounds / Gallon Product Name Column A Column B Acetylene UN/NA: 1001 14.70 4.90 CAS: 514-86-2 Air UN/NA: 1002 13.30 7.29 CAS: N/A Ammonia Anhydrous UN/NA: 1005 20.78 5.147 CAS: 7664-41-7 Argon UN/NA: 1006 9.71 11.63 CAS: 7440-37-1 Butane UN/NA: 1075 6.34 4.86 CAS: 106-97-8 Carbon Dioxide UN/NA: 2187 8.74 8.46 CAS: 124-38-9 Chlorine UN/NA: 1017 5.38 11.73 CAS: 7782-50-5 Ethane UN/NA: 1045 12.51 2.74 CAS: 74-84-0 Ethylene Oxide UN/NA: 1040 8.78 7.25 CAS: 75-21-8 Fluorine UN/NA: 1045 10.17 12.60 CAS: 7782-41-4 Helium UN/NA: 1046 97.09 1.043 CAS: 7440-59-7 Hydrogen UN/NA: 1049 192.00 0.592 CAS: 1333-74-0 1. Find the gas you want to convert. 2. If you know your quantity in cubic feet and want to convert to pounds, divide your amount by column A 3. If you know your quantity in gallons and want to convert to pounds, multiply your amount by column B 4. If you know your quantity in pounds and want to convert to gallons, divide your amount by column B If you have any questions, please call 1-800-433-2288 LIQUEFIED GAS CONVERSION CHART Cubic Feet / Pound Pounds / Gallon Product Name Column A Column B Hydrogen Chloride UN/NA: 1050 10.60 8.35 CAS: 7647-01-0 Krypton UN/NA: 1056 4.60 20.15 CAS: 7439-90-9 Methane UN/NA: 1971 23.61 3.55 CAS: 74-82-8 Methyl Bromide UN/NA: 1062 4.03 5.37 CAS: 74-83-9 Neon UN/NA: 1065 19.18 10.07 CAS: 7440-01-9 Mapp Gas UN/NA: 1060 9.20 4.80 CAS: N/A Nitrogen UN/NA: 1066 13.89 6.75 CAS: 7727-37-9 Nitrous Oxide UN/NA: 1070 8.73 6.45 CAS: 10024-97-2 Oxygen UN/NA: 1072 12.05 9.52 CAS: 7782-44-7 Propane UN/NA: 1075 8.45 4.22 CAS: 74-98-6 Sulfur Dioxide UN/NA: 1079 5.94 12.0 CAS: 7446-09-5 Xenon UN/NA: 2036 2.93 25.51 CAS: 7440-63-3 1.