A De Novo Microdeletion of SEMA5A in a Boy with Autism Spectrum Disorder and Intellectual Disability
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Gene Ontology Enrichment Analysis in Two Independent Family-Based
Gene ontology enrichment analysis in two independent family-based samples highlights biologically plausible processes for autism spectrum disorders Richard Jl Anney, Elizabeth A Heron, Ricardo Segurado, Elaine M Kenny, Colm O’Dushlaine, Brian L Yaspan, Elena Parkhomenko, The Autism Genome Project, Joseph Buxbaum, James S Sutcliffe, et al. To cite this version: Richard Jl Anney, Elizabeth A Heron, Ricardo Segurado, Elaine M Kenny, Colm O’Dushlaine, et al.. Gene ontology enrichment analysis in two independent family-based samples highlights biologically plausible processes for autism spectrum disorders. European Journal of Human Genetics, Nature Publishing Group, 2011, 10.1038/ejhg.2011.75. hal-00636189 HAL Id: hal-00636189 https://hal.archives-ouvertes.fr/hal-00636189 Submitted on 27 Oct 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Title Page Gene ontology enrichment analysis in two independent family-based samples highlights biologically plausible processes for autism spectrum disorders Running Title Gene pathways analysis in ASD Word Count 158 (Abstract) 3491 (Manuscript Body) Author List Richard J.L. Anney 1†PhD, Elizabeth A. Heron 1 PhD, Ricardo Segurado 1 PhD, Elaine M. Kenny 1 PhD, Colm O'Dushlaine 1,2 PhD, Brian L. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
H Supplemental Figure 1 I D E B a C
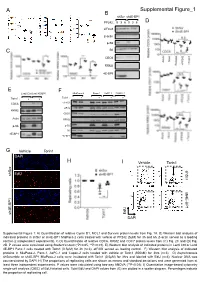
A Supplemental Figure_1 B shScr sh4E-BP1 PP242 : 0 3 6 0 3 6 D eIF4G1 β-actin p-S6 C S6 CDC6 RRM2 4E-BP1 E F Lenti Ctrl Lenti 4E-BP1 MiaPaca-2 Panc-1 AsPC-1 Capan-2 Torin1 : Torin1 : + + + + + + CDC6 eIF4G1 eIF3a RRM2 CDC6 Actin RRM2 p-S6 p-S6 S6 4E-BP1 4E-BP1 G Vehicle Torin1 DAPI H I Vehicle Torin1 62.20% 45.30% shScr sh4E-BP1 EdU shScr 59.58% 49.47% EdU sh4E-BP1 DAPI Supplemental Figure 1: A) Quantification of relative Cyclin D1, MCL1 and Survivin protein levels from Fig. 1A. B) Western blot analysis of indicated proteins in shScr or sh4E-BP1 MiaPaca-2 cells treated with vehicle or PP242 (5µM) for 3h and 6h. β-actin served as a loading control (2 independent experiments). C-D) Quantification of relative CDC6, RRM2 and CDC7 protein levels from (C) Fig. 2C and (D) Fig. 2D. P values were calculated using Student’s t-test (*P<0.05, **P<0.01). E) Western blot analysis of indicated proteins in Lenti Ctrl or Lenti 4E-BP1 Panc-1 cells treated with Torin1 (0.5µM) for 3h (n=3). eIF4GI served as loading control. F) Western blot analysis of indicated proteins in MiaPaca-2, Panc-1, AsPC-1 and Capan-2 cells treated with vehicle or Torin1 (500nM) for 3hrs (n=3). G) Asynchronous shScramble or sh4E-BP1 MiaPaca-2 cells were incubated with Torin1 (0.5μM) for 3hrs and labeled with EdU (n=3). Nuclear DNA was counterstained by DAPI. H) The proportions of replicating cells are shown as means and standard deviations and were generated from at least three independent experiments. -

Multivariate Analysis Reveals Genetic Associations of the Resting Default
Multivariate analysis reveals genetic associations of PNAS PLUS the resting default mode network in psychotic bipolar disorder and schizophrenia Shashwath A. Medaa,1, Gualberto Ruañob,c, Andreas Windemuthb, Kasey O’Neila, Clifton Berwisea, Sabra M. Dunna, Leah E. Boccaccioa, Balaji Narayanana, Mohan Kocherlab, Emma Sprootena, Matcheri S. Keshavand, Carol A. Tammingae, John A. Sweeneye, Brett A. Clementzf, Vince D. Calhoung,h,i, and Godfrey D. Pearlsona,h,j aOlin Neuropsychiatry Research Center, Institute of Living at Hartford Hospital, Hartford, CT 06102; bGenomas Inc., Hartford, CT 06102; cGenetics Research Center, Hartford Hospital, Hartford, CT 06102; dDepartment of Psychiatry, Beth Israel Deaconess Hospital, Harvard Medical School, Boston, MA 02215; eDepartment of Psychiatry, University of Texas Southwestern Medical Center, Dallas, TX 75390; fDepartment of Psychology, University of Georgia, Athens, GA 30602; gThe Mind Research Network, Albuquerque, NM 87106; Departments of hPsychiatry and jNeurobiology, Yale University, New Haven, CT 06520; and iDepartment of Electrical and Computer Engineering, The University of New Mexico, Albuquerque, NM 87106 Edited by Robert Desimone, Massachusetts Institute of Technology, Cambridge, MA, and approved April 4, 2014 (received for review July 15, 2013) The brain’s default mode network (DMN) is highly heritable and is Although risk for psychotic illnesses is driven in small part by compromised in a variety of psychiatric disorders. However, ge- highly penetrant, often private mutations such as copy number netic control over the DMN in schizophrenia (SZ) and psychotic variants, substantial risk also is likely conferred by multiple genes bipolar disorder (PBP) is largely unknown. Study subjects (n = of small effect sizes interacting together (7). According to the 1,305) underwent a resting-state functional MRI scan and were “common disease common variant” (CDCV) model, one would analyzed by a two-stage approach. -

Analysis of Gene Expression in the Nervous System Identifies Key Genes and Novel Candidates for Health and Disease
Neurogenetics (2017) 18:81–95 DOI 10.1007/s10048-017-0509-5 ORIGINAL ARTICLE Analysis of gene expression in the nervous system identifies key genes and novel candidates for health and disease Sarah M Carpanini1 & Thomas M Wishart1 & Thomas H Gillingwater2 & Jean C Manson1 & Kim M Summers1 Received: 13 July 2016 /Accepted: 20 January 2017 /Published online: 11 February 2017 # The Author(s) 2017. This article is published with open access at Springerlink.com Abstract The incidence of neurodegenerative diseases in the pathways and gene ontology term annotation. Additionally a developed world has risen over the last century, concomitant number of poorly annotated genes were implicated by this with an increase in average human lifespan. A major chal- approach in nervous system function. Exploiting gene expres- lenge is therefore to identify genes that control neuronal health sion data available in the public domain allowed us to validate and viability with a view to enhancing neuronal health during key nervous system genes and, importantly, to identify additional ageing and reducing the burden of neurodegeneration. genes with minimal functional annotation but with the same Analysis of gene expression data has recently been used to expression pattern. These genes are thus novel candidates for infer gene functions for a range of tissues from co-expression a role in neurological health and disease and could now be networks. We have now applied this approach to further investigated to confirm their function and regulation transcriptomic datasets from the mammalian nervous system during ageing and neurodegeneration. available in the public domain. We have defined the genes critical for influencing neuronal health and disease in different Keywords Mice . -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Whole Exome Sequencing Reveals a Novel AUTS2 In-Frame Deletion in a Boy with Global Developmental Delay, Absent Speech, Dysmorphic Features, and Cerebral Anomalies
G C A T T A C G G C A T genes Case Report Whole Exome Sequencing Reveals a Novel AUTS2 In-Frame Deletion in a Boy with Global Developmental Delay, Absent Speech, Dysmorphic Features, and Cerebral Anomalies Pietro Palumbo 1 , Ester Di Muro 1, Maria Accadia 2, Mario Benvenuto 1, Marilena Carmela Di Giacomo 3, Stefano Castellana 4, Tommaso Mazza 4 , Marco Castori 1, Orazio Palumbo 1,* and Massimo Carella 1 1 Division of Medical Genetics, Fondazione IRCCS-Casa Sollievo della Sofferenza, 71013 San Giovanni Rotondo (Foggia), Italy; [email protected] (P.P.); [email protected] (E.D.M.); [email protected] (M.B.); [email protected] (M.C.); [email protected] (M.C.) 2 Medical Genetics Service, Hospital “Cardinale G. Panico”, 73039 Tricase (Lecce), Italy; [email protected] 3 U.O.C di Anatomia Patologica, AOR Ospedale “San Carlo”, 85100 Potenza, Italy; [email protected] 4 Unit of Bioinformatics, Fondazione IRCCS Casa Sollievo della Sofferenza, 71013 San Giovanni Rotondo (Foggia), Italy; [email protected] (S.C.); [email protected] (T.M.) * Correspondence: [email protected] Abstract: Neurodevelopmental disorders (NDDs) are a group of highly prevalent, clinically and genetically heterogeneous pediatric disorders comprising, according to the Diagnostic and Statisti- Citation: Palumbo, P.; Di Muro, E.; cal Manual of Mental Disorders 5th edition (DSM-V), intellectual disability, developmental delay, Accadia, M.; Benvenuto, M.; Di autism spectrum disorders, and other neurological and cognitive disorders manifesting in the de- Giacomo, M.C.; Castellana, S.; Mazza, velopmental age. To date, more than 1000 genes have been implicated in the etiopathogenesis of T.; Castori, M.; Palumbo, O.; Carella, NNDs. -

Constitutional Downregulation of SEMA5A Expression in Autism
Constitutional downregulation of SEMA5A expression in autism. Malin Melin, Birgit Carlsson, Henrik Anckarsäter, Maria Rastam, Catalina Betancur, Anders Isaksson, Christopher Gillberg, Niklas Dahl To cite this version: Malin Melin, Birgit Carlsson, Henrik Anckarsäter, Maria Rastam, Catalina Betancur, et al.. Con- stitutional downregulation of SEMA5A expression in autism.. Neuropsychobiology, Karger, 2006, 54 (1), pp.64-9. 10.1159/000096040. inserm-00124743 HAL Id: inserm-00124743 https://www.hal.inserm.fr/inserm-00124743 Submitted on 2 Sep 2008 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Melin et al. 1 Constitutional down-regulation of SEMA5A expression in autism M. Melina, B. Carlssona, H. Anckarsaterb, M. Rastamb, C. Betancurc, A. Isakssona, C. Gillbergb, N. Dahla aDepartment of Genetics and Pathology, Uppsala University, Uppsala, Sweden bDepartment of Child and Adolescent Psychiatry, Göteborg University, Göteborg, Sweden cINSERM U513, Université Paris XII, Créteil, France Correspondence should be addressed to Professor Niklas Dahl, Uppsala University, Department of Genetics and Pathology, Rudbeck Laboratory, 751 85 Uppsala, Sweden; e- mail: [email protected] Melin et al. 2 Abstract There is strong evidence for the importance of genetic factors in idiopathic autism. The results from independent twin and family studies suggest that the disorder is caused by the action of several genes, possibly acting epistatically. -

The Role of Semaphorin 5A in Pancreatic Cancer Progression and Metastasis
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Spring 5-6-2017 The Role of Semaphorin 5A in Pancreatic Cancer Progression and Metastasis Sugandha Saxena Dr. University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Biochemistry Commons, and the Cancer Biology Commons Recommended Citation Saxena, Sugandha Dr., "The Role of Semaphorin 5A in Pancreatic Cancer Progression and Metastasis" (2017). Theses & Dissertations. 200. https://digitalcommons.unmc.edu/etd/200 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. THE ROLE OF SEMAPHORIN 5A IN PANCREATIC CANCER PROGRESSION AND METASTASIS By Sugandha Saxena A DISSERTATION Presented to the Faculty of The Graduate College of the University of Nebraska Medical Center In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor Rakesh K. Singh University of Nebraska Medical Center Omaha, Nebraska April 2017 Supervisory Committee Surinder K. Batra, Ph.D. Joyce Solheim, Ph.D. Micheal A. (Tony) Hollingsworth, Ph.D. Michel Ouellette, Ph.D. i THE ROLE OF SEMAPHORIN 5A IN PANCREATIC CANCER PROGRESSION AND METASTASIS Sugandha Saxena, Ph.D. University of Nebraska Medical Center Supervisor: Rakesh Singh, Ph.D. This dissertation investigates the pathological significance and functional role of Semaphorin 5A (SEMA5A), an axon guidance molecule, during Pancreatic Cancer (PC) progression and metastasis. -

Cri-Du-Chat Syndrome Diagnosed in a 21-Year-Old Woman by Means of Comparative Genomic Hybridization
Rev. Fac. Med. 2017 Vol. 65 No. 3: 525-9 525 CASE REPORT DOI: http://dx.doi.org/10.15446/revfacmed.v65n3.57414 Cri-du-chat syndrome diagnosed in a 21-year-old woman by means of comparative genomic hybridization Síndrome de cri du chat diagnosticado en mujer de 21 años por hibridación genómica comparativa Received: 15/05/2016. Accepted: 06/08/2016. Wilmar Saldarriaga1 • Laura Collazos-Saa2 • Julián Ramírez-Cheyne2 1 Universidad del Valle - Faculty of Health - Department Morphology - MACOS Research Group - Cali - Colombia. 2 Universidad del Valle - Faculty of Health - Medicine and Surgery - Cali - Colombia. Corresponding author: Wilmar Saldarriaga. Department Morphology, Faculty of Health, Universidad del Valle. Calle 4B No. 36-00, building 116, floor 1, espacio 30. Phone number: +57 2 5285630, ext.: 4030; mobile number: +57 3164461596. Cali. Colombia. Email: [email protected]. | Abstract | entero. La prevalencia va desde 1 por 15 000 habitantes hasta 1 por 50 000 habitantes. Su diagnóstico se puede confirmar con cariotipo The cri-du-chat syndrome is caused by a deletion on the short con bandas G de alta resolución, hibridación fluorescente in situ o arm of chromosome number 5. The size of genetic material loss hibridación genómica comparativa por microarreglos (HGCm); este varies from the 5p15.2 region only to the whole arm. Prevalence se sospecha en infantes con un llanto similar al maullido de un gato, rates range between 1:15000 and 1:50000 live births. Diagnosis fascies dismórficas, hipotonía y retardo del desarrollo psicomotor; is suspected on infants with a high-pitched (cat-like) cry, facial sin embargo, en los adultos afectados los hallazgos fenotípicos son dysmorfism, hypotonia and delayed psychomotor development. -

Identification of Semaphorin 5A Interacting Protein by Applying Apriori Knowledge and Peptide Complementarity Related to Protein Evolution and Structure
Article Identification of Semaphorin 5A Interacting Protein by Applying Apriori Knowledge and Peptide Complementarity Related to Protein Evolution and Structure Anguraj Sadanandam, Michelle L. Varney, and Rakesh K. Singh* Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198-5845, USA. In the post-genomic era, various computational methods that predict protein- protein interactions at the genome level are available; however, each method has its own advantages and disadvantages, resulting in false predictions. Here we devel- oped a unique integrated approach to identify interacting partner(s) of Semaphorin 5A (SEMA5A), beginning with seven proteins sharing similar ligand interacting residues as putative binding partners. The methods include Dwyer and Root- Bernstein/Dillon theories of protein evolution, hydropathic complementarity of protein structure, pattern of protein functions among molecules, information on domain-domain interactions, co-expression of genes and protein evolution. Among the set of seven proteins selected as putative SEMA5A interacting partners, we found the functions of Plexin B3 and Neuropilin-2 to be associated with SEMA5A. We modeled the semaphorin domain structure of Plexin B3 and found that it shares similarity with SEMA5A. Moreover, a virtual expression database search and RT-PCR analysis showed co-expression of SEMA5A and Plexin B3 and these proteins were found to have co-evolved. In addition, we confirmed the interac- tion of SEMA5A with Plexin B3 in co-immunoprecipitation studies. Overall, these studies demonstrate that an integrated method of prediction can be used at the genome level for discovering many unknown protein binding partners with known ligand binding domains. Key words: domain-domain interaction, semaphorin, plexin, protein interaction prediction Introduction Protein-protein interactions play a major role dur- of a coat protein gene on the surface of bacterio- ing normal cellular functions or pathological diseases. -

Integrative Analysis of Disease Signatures Shows Inflammation Disrupts Juvenile Experience-Dependent Cortical Plasticity
New Research Development Integrative Analysis of Disease Signatures Shows Inflammation Disrupts Juvenile Experience- Dependent Cortical Plasticity Milo R. Smith1,2,3,4,5,6,7,8, Poromendro Burman1,3,4,5,8, Masato Sadahiro1,3,4,5,6,8, Brian A. Kidd,2,7 Joel T. Dudley,2,7 and Hirofumi Morishita1,3,4,5,8 DOI:http://dx.doi.org/10.1523/ENEURO.0240-16.2016 1Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 2Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 3Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 4Department of Ophthalmology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 5Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 6Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 7Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, and 8Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029 Visual Abstract Throughout childhood and adolescence, periods of heightened neuroplasticity are critical for the development of healthy brain function and behavior. Given the high prevalence of neurodevelopmental disorders, such as autism, identifying disruptors of developmental plasticity represents an essential step for developing strategies for prevention and intervention. Applying a novel computational approach that systematically assessed connections between 436 transcriptional signatures of disease and multiple signatures of neuroplasticity, we identified inflammation as a common pathological process central to a diverse set of diseases predicted to dysregulate Significance Statement During childhood and adolescence, heightened neuroplasticity allows the brain to reorganize and adapt to its environment.