Fusing Nickelocene and Cyclopentadiene by Two Silyl Bridges
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Application of Metallocenes for the Synthesis of Multiwalled Carbon Nanotubes
APPLICATION OF METALLOCENES FOR THE SYNTHESIS OF MULTIWALLED CARBON NANOTUBES by Nonjabulo Philile Dominica Ngidi Submitted in fulfilment of the academic requirement for the degree of Master of Science in the School of Chemistry and Physics University of KwaZulu-Natal, Durban, Westville campus February 2016 ABSTRACT Multiwalled carbon nanotubes (MWCNTs) are carbon materials which have one-dimensional structure. They possess unique properties such as semi-conductor and high tensile strength that allow them to be widely used in many applications. MWCNTs and other shaped carbon nanomaterials (SCNMs) were synthesized by chemical vapour deposition (CVD) method. Three factors that affect the morphology, thermal, chemical, mechanical and electrical properties of SCNMs were investigated. The parameters are: carbon source, catalyst (metallocenes), and growth temperature. Two different carbon sources were studied for the synthesis of MWCNTs i.e., toluene and acetonitrile (also used as a nitrogen source). The metallocenes: nickelocene, cobaltocene and ruthenocene were used as catalysts (2.5 wt.%) while ferrocene was employed as control. These metallocenes were investigated because they have similar structure as ferrocene, a well-known catalyst for the synthesis of SCNMs. The synthesis was carried out at five different growth temperatures, 800, 850, 900, 950 and 1000 °C. As-synthesized MWCNTs and other SCNMs were further purified, in order to remove amorphous carbon and this was performed by testing different methods of purification. The effective method for purification of MWCNTs was method 2 which involved refluxing for 24 hours. It was chosen to be the best method because it produced purer MWCNTs as compared to other methods which caused damage to the MWCNTs. -

Edinburgh Research Explorer
Edinburgh Research Explorer Organometallic Neptunium Chemistry Citation for published version: Arnold, P, Dutkiewicz, MS & Walter, O 2017, 'Organometallic Neptunium Chemistry', Chemical Reviews. https://doi.org/10.1021/acs.chemrev.7b00192 Digital Object Identifier (DOI): 10.1021/acs.chemrev.7b00192 Link: Link to publication record in Edinburgh Research Explorer Document Version: Peer reviewed version Published In: Chemical Reviews General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 29. Sep. 2021 Organometallic Neptunium Chemistry Polly L. Arnold,*a Michał S. Dutkiewicz,a,b Olaf Walter,b [a] EaStCHEM School of Chemistry, University of Edinburgh, The King’s Buildings, Edinburgh, EH9 3FJ, UK. E-mail: [email protected]. [b] European Commission, DG Joint Research Centre, Directorate G - Nuclear Safety and Security, Advanced Nuclear Knowledge – G.I.5, Postfach 2340, D-76125, Karlsruhe, Germany. ABSTRACT Fifty years have passed since the foundation of organometallic neptunium chemistry, and yet only a handful of complexes have been reported, and even fewer fully characterised. Yet increasingly, combined synthetic/spectroscopic/computational studies are demonstrating how covalently binding, soft, carbocyclic organometallic ligands provide an excellent platform for advancing the fundamental understanding of the differences in orbital contributions and covalency in f-block metal – ligand bonding. -

Synthesis and Reactivity of Cyclopentadienyl Based Organometallic Compounds and Their Electrochemical and Biological Properties
Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Sasmita Mishra Department of Chemistry National Institute of Technology Rourkela Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Dissertation submitted to the National Institute of Technology Rourkela In partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry by Sasmita Mishra (Roll Number: 511CY604) Under the supervision of Prof. Saurav Chatterjee February, 2017 Department of Chemistry National Institute of Technology Rourkela Department of Chemistry National Institute of Technology Rourkela Certificate of Examination Roll Number: 511CY604 Name: Sasmita Mishra Title of Dissertation: ''Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties We the below signed, after checking the dissertation mentioned above and the official record book(s) of the student, hereby state our approval of the dissertation submitted in partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry at National Institute of Technology Rourkela. We are satisfied with the volume, quality, correctness, and originality of the work. --------------------------- Prof. Saurav Chatterjee Principal Supervisor --------------------------- --------------------------- Prof. A. Sahoo. Prof. G. Hota Member (DSC) Member (DSC) --------------------------- -

1 Signature Page Biphenyl Substituted Cyclopentadienyl Ligand
Signature page Biphenyl substituted Cyclopentadienyl Ligand Complexes By Ketnavi Ramgoolam A Thesis Submitted to Saint Mary’s University, Halifax, Nova Scotia in Partial Fulfillment of the Requirements for the Degree of Bachelor of Science Degree with Honours in Chemistry April 2019, Halifax, Nova Scotia Copyright Ketnavi Ramgoolam, 2019. Approved: Dr. Jason Masuda Supervisor Department of Chemistry Date: April 24, 2019 1 Biphenyl substituted Cyclopentadienyl Ligand Complexes By Ketnavi Ramgoolam A Thesis Submitted to Saint Mary’s University, Halifax, Nova Scotia in Partial Fulfillment of the Requirements for the Degree of Bachelor of Science Degree with Honours in Chemistry April 2019, Halifax, Nova Scotia Copyright Ketnavi Ramgoolam, 2019. Approved: Dr. Jason Masuda Supervisor Department of Chemistry Date: April 24, 2019 2 Biphenyl substituted Cyclopentadienyl Ligand Complexes by Ketnavi Ramgoolam Abstract The synthesis of biphenyl substituted cyclopentadienyl ligands are described. The synthetic route is accomplished through the reaction of aryl lithium reagents with cobaltocenium salts, followed by oxidation of the intermediate cobalt(I) species to give the corresponding cyclopentadiene. Detailed information on their preparation, structural, and spectroscopic properties are described. A preliminary reaction towards the biphenyl substituted cyclopentadienyl ligand complexes yielding the corresponding alkali metal salt (K) is also described. Its structural and spectroscopic properties are described briefly. March 20, 2019 3 Acknowledgements First of all, I would like to express my most sincere gratitude to my supervisor, Dr. Jason Masuda, for all of his time, efforts, teachings and guidance. Thank you for being a most wonderful and understanding human being. This project helped me in discovering my potential as a chemist and I had plenty of wonderful experiences and opportunities that I personally believe will forever shape and influence my professional life as well as my personal growth. -

Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005)
NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005 IUPAC Periodic Table of the Elements 118 1 2 21314151617 H He 3 4 5 6 7 8 9 10 Li Be B C N O F Ne 11 12 13 14 15 16 17 18 3456 78910 11 12 Na Mg Al Si P S Cl Ar 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 55 56 * 57− 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba lanthanoids Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 87 88 ‡ 89− 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 Fr Ra actinoids Rf Db Sg Bh Hs Mt Ds Rg Uub Uut Uuq Uup Uuh Uus Uuo * 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu ‡ 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr International Union of Pure and Applied Chemistry Nomenclature of Inorganic Chemistry IUPAC RECOMMENDATIONS 2005 Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry Prepared for publication by Neil G. -

CURRICULUM VITAE WILLIAM E. GEIGER, JR. Professor Of
CURRICULUM VITAE WILLIAM E. GEIGER, JR. Professor of Chemistry University of Vermont, Burlington VT 05405 802-656-0268; EMAIL [email protected] FAX 802-656-8705 Education B.S. Canisius College 1965 Chemistry Ph.D. Cornell University 1969 Analytical Chemistry Positions Research Associate, Univ. of California at Riverside 1968-69 Postdoctoral Research Associate, Northwestern Univ. 1969-70 Assistant Professor, Southern Illinois Univ. 1970-74 Assistant Professor, Univ. of Vermont 1974-77 Associate Professor, Univ. of Vermont 1977-82 Professor, Univ. of Vermont 1982-Present Pomeroy Professor, Univ. of Vermont 1997-Present Chair, Department of Chemistry, University of Vermont 1998-2001 Honors University of Vermont Scholar 1984 James Crowdle Award, Canisius College 1995 Dean’s Lecturer, UVM 2009 Vermont Academy of Science and Engineering 2009 William Geiger Research Laboratory (Free State University) 2011 Professional Organizations Member, American Chemical Society (ACS) Division of Analytical Chemistry Division of Inorganic Chemistry Society for Electroanalytical Chemistry (SEAC) Vermont Academy of Science and Engineering Selected Professional Activities Professeur Associe, Univ. of Bordeaux 1986 Editor, Newsletter of SEAC 1985-88 Editorial Advisory Board, Organometallics 1989-91 Board of Directors, SEAC 1992-95 Graduiertenkolleg Lecturer, Univ. of Freiburg 1996 Editorial Board, Chemtracts 1997-98 Leverhulme Fellow, University of Bristol 1998 Erskine Fellow, University of Canterbury 2000 Advisory Board, Center for Molecular Electrocatalysis 2009-present INVITED LECTURES AND COLLOQUIA Canisius College 10/70 Univ. Maryland at Baltimore 9/71 Franklin and Marshall College 9/71 Univ. of Missouri at St. Louis 2/73 Brown University 10/75 Univ. of Massachusetts at Amherst 12/75 Univ. of Massachusetts at Boston 2/76 SUNY at Buffalo 11/76 SUNY at Plattsburgh 1/77 Providence College 2/78 Wesleyan University 2/78 St. -

Synthesis of the Metallocenes for the Production of Exotic High Energy Ion Beams
Synthesis of the Metallocenes for the Production of Exotic High Energy Ion Beams Ntombizonke Yvonne Kheswa A thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the Department of Physics & Astronomy, University of the Western Cape, South Africa. Supervised by: Prof. J. N. Orce, Department of Physics & Astronomy University of the Western Cape Prof. S. Titinchi, Department of Chemistry, University of the Western Cape Dr. R. Thomae Accelerator and Engineering Department iThemba LABS March 2019 https://etd.uwc.ac.za DECLARATION I declare that Synthesis of the Metallocenes for the Production of Exotic High Energy Ion Beams is my own work, that it has not been submitted for any degree or examination in any other university, and that all the sources I have used or quoted have been indicated and acknowledged by complete references. Signed: Ntombizonke Kheswa Date: 1 March 2019 i https://etd.uwc.ac.za Synthesis of the Metallocenes for the Production of Exotic High Energy Ion Beams Department of Physics and Astronomy, University of the Western Cape, Private Bag X17, 7535 Bellville, South Africa. ABSTRACT The Subatomic Physics Department of iThemba Laboratory for Accelerated Based Sciences (iThemba LABS) conducts experiments that require a variety of particle beams in order to study nuclear properties (reaction, structure, etc.) of various nuclides. These particle beams are accelerated using the K-200 Separated Sector Cyclotron (SSC) and delivered to different physics experimental vaults. Prior to acceleration, the particle beam is first ionised using an Electron Resonance Ion Source (ECRIS). The main goal of this study is the production of exotic metallic beams of 60Ni8+ and 62Ni8+ using ECRIS4, which are required for the Coulomb excitation experiments approved by the Programme Advisory Committee (PAC) at iThemba LABS. -

Differentiation Between Spin State, Electrostatic and Covalent Bonding
Inorganica Chimica Acta 360 (2007) 179–189 www.elsevier.com/locate/ica Metal–ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding Marcel Swart Institucio´ Catalana de Recerca i Estudis Avanc¸ats (ICREA), 08010 Barcelona, Spain Institut de Quı´mica Computacional, Universitat de Girona, Campus Montilivi, 17071 Girona, Spain Received 16 June 2006; received in revised form 26 July 2006; accepted 26 July 2006 Available online 5 August 2006 Inorganic Chemistry – The Next Generation. Abstract We have analyzed metal–ligand bonding in metallocenes using density functional theory (DFT) at the OPBE/TZP level. This level of theory was recently shown to be the only DFT method able to correctly predict the spin ground state of iron complexes, and similar accuracy for spin ground states is found here. We considered metallocenes along the first-row transition metals (Sc–Zn) extended with alkaline-earth metals (Mg, Ca) and several second-row transition metals (Ru, Pd, Ag, Cd). Using an energy decomposition analysis, we have studied trends in metal–ligand bonding in these complexes. The OPBE/TZP enthalpy of heterolytic association for ferrocene (À658 kcal/mol) as obtained from the decomposition analysis is in excellent agreement with benchmark CCSD(T) and CASPT2 results. Covalent bonding is shown to vary largely for the different metallocenes and is found in the range from À155 to À635 kcal/mol. Much smaller variation is observed for Pauli repulsion (55–345 kcal/mol) or electrostatic interactions, which are however strong (À480 to À620 kcal/mol). The covalent bonding, and thus the metal–ligand bonding, is larger for low spin states than for higher spin states, due to better suitability of acceptor d-orbitals of the metal in the low spin state. -

N-Metal Compounds
31 N-METAL COMPOUNDS n the 85 years following Kekulk's brilliant proposal for the structure of ben- zene, organic chemistry underwent a tremendous expansion, and in the process a wide variety of paradigms or working hypotheses were developed about what kinds of compounds could "exist" and what kinds of reactions could occur. In many cases, acceptance of these hypotheses appeared to stifle many possible lines of investigation and caused contrary evidence to be pigeonholed as "interesting but not conclusive." As one example, the paradigm of angle strain was believed to wholly preclude substances that we know now are either stable or important reaction intermediates, such as cubane (Section 12- 1O), cyclo- propanone (Section 17-1 I), and benzyne (Sections 14-6C and 23-8). No paradigm did more to retard the development of organic chemistry than the notion that, with a "few" exceptions, compounds with bonds between carbon and transition metals (Fe, Co, Ni, Ti, and so on) are inherently un- stable. This idea was swept away in 1951 with the discovery of ferrocene, (C,H,),Fe, by P. L. Pauson. Ferrocene has unheard of properties for an organoiron compound, stable to more than 500" and able to be dissolved in, and recovered from, concentrated sulfuric acid! Pauson's work started an avalanche of research on transition metals in the general area between organic and inorganic chemistry, which has flourished ever since and has led to an improved understanding of important biochemical processes. 31-1 Metallocenes 31-1 METALLOCENES The discovery of ferrocene was one of those fortuitous accidents that was wholly unforeseeable- the kind of discovery which, over and over again, has changed the course of science. -

"Mixed-Ligand" Homocyclopentadienyl Ruthenocenes
Western Michigan University ScholarWorks at WMU Master's Theses Graduate College 12-1992 Synthesis of Symmetric and "Mixed-Ligand" Homocyclopentadienyl Ruthenocenes John Adjeiku Amanfu Follow this and additional works at: https://scholarworks.wmich.edu/masters_theses Part of the Organic Chemistry Commons Recommended Citation Amanfu, John Adjeiku, "Synthesis of Symmetric and "Mixed-Ligand" Homocyclopentadienyl Ruthenocenes" (1992). Master's Theses. 902. https://scholarworks.wmich.edu/masters_theses/902 This Masters Thesis-Open Access is brought to you for free and open access by the Graduate College at ScholarWorks at WMU. It has been accepted for inclusion in Master's Theses by an authorized administrator of ScholarWorks at WMU. For more information, please contact [email protected]. SYNTHESIS OF SYMMETRIC AND "MDCED-LIGAND" HOMOCYCLOPENTADEENYL RUTHENOCENES John Adjeiku Amanfu A Thesis Submitted to the Faculty of The Graduate College in partial fulfillment of the requirements for the Degree of Master of Arts Department of Chemistry Western Michigan University Kalamazoo, Michigan December 1992 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. SYNTHESIS OF SYMMETRIC AND "MIXED-LIGAND HOMOCYCLOPENTADIENYL RUTHENOCENES John Adjeiku Amanfu, M.A. Western Michigan University, 1992 Muller and co-workers were the first to report the synthesis of a symmetric bicyclic analogue of ferroene, by treating the anion of bicy- cio[3.2.2]nona-2,6,8-triene (BCNT) with ferrous chloride. This compound was found to be thermally less stable than ferrocene decomposing above 50°C. We report the synthesis and characterization of two homocy- clopentadienyl ruthenocenes; namely (pentamethylcyclopentadienyl)- (bicyclo[3.2.1]octa-2,6-dienyl) ruthenium(II), {ri^-(C5Me5)Ru(BCOD)} and (cyclopentadienyl)(bicyclo[3.2.1]octa-2,6-dienyl)ruthenium(II) {^-(CsHs)- Ru(BCOD)} by treating bicyclo[3.2.1]octa-2,6-dienyl anion with the ruthenium complexes, [Cp*RuCl2]2 and [(NBD)RuCl 2 ]2 respectively. -

Electrochemistry of Metallocenes at Very Negative and Very Positive Potentials
Subscriber access provided by Univ. of Texas Libraries Electrochemistry of metallocenes at very negative and very positive potentials. Electrogeneration of 17-electron Cp2Co2+, 21-electron Cp2Co2-, and 22-electron Cp2Ni2- species Allen J. Bard, Edwin Garcia, S. Kukharenko, and Vladimir V. Strelets Inorg. Chem., 1993, 32 (16), 3528-3531 • DOI: 10.1021/ic00068a024 Downloaded from http://pubs.acs.org on January 23, 2009 More About This Article The permalink http://dx.doi.org/10.1021/ic00068a024 provides access to: • Links to articles and content related to this article • Copyright permission to reproduce figures and/or text from this article Inorganic Chemistry is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 352a Inorg. Chem. 1993, 32, 3528-3531 Electrochemistry of Metallocenes at Very Negative and Very Positive Potentials. Electrogeneration of 17-Electron Cp2C02+, Cp2C02-, and Cp2Ni2- Species Allen J. Bard' and Edwin Garcia Department of Chemistry and Biochemistry, The University of Texas at Austin, Austin, Texas 78712 S. Kukbarenko and Vladimir V. Strelets' Institute of Chemical Physics, Russian Academy of Sciences, Chernogolovka, Moscow Region 142432, Russia Received January 14, 1993 Cyclic voltammetry (CV) with Pt ultramicroelectrodes and coulometry were used to study the electrooxidation of the Cp2Co+ cation in liquid S02/(TBA)AsFs solution in the temperature range from -70 to +25 OC. The Cp2Co+ cation was shown to undergo reversible one-electron oxidation with formation of the Cp2Co2+ dication, which is stable on the CV time scale. The chemical and electrochemical reversibility of CpzCo+ oxidation suggests retention of the sandwich structure in the dication. -
A Qualitative Molecular Orbital Diagram for Ferrocene
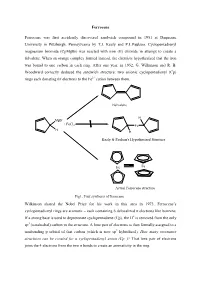
Ferrocene Ferrocene was first accidently discovered sandwich compound in 1951 at Duquesne University in Pittsburgh, Pennsylvania by T.J. Kealy and P.L.Paulson. Cyclopentadienyl magnesium bromide (CpMgBr) was reacted with iron (II) chloride in attempt to create a fulvalene. When an orange complex formed instead, the chemists hypothesized that the iron was bound to one carbon in each ring. After one year, in 1952, G. Wilkinson and R. B. Woodward correctly deduced the sandwich structure: two anionic cyclopentadienyl (Cp) rings each donating 6π electrons to the Fe2+ cation between them. Fulvalene H MgBr + FeCl2 Fe H H Kealy & Paulson's Hypothesized Structure Fe Fe Actual Ferrocene structure Fig1. First synthesis of ferrocene Wilkinson shared the Nobel Prize for his work in this area in 1973. Ferrocene’s cyclopentadienyl rings are aromatic – each containing 6 delocalized π electrons like benzene. If a strong base is used to deprotonate cyclopentadiene (Cp), the H+ is removed from the only sp3 (tetrahedral) carbon in the structure. A lone pair of electrons is then formally assigned to a nonbonding p orbital of that carbon (which is now sp2 hybridized). How many resonance structures can be created for a cyclopentadienyl anion (Cp−)? That lone pair of electrons joins the 4 electrons from the two π bonds to create an aromaticity in the ring. :B H H H : Fig 2. Aromaticity of cyclopentadienyl ring Properties of Ferrocene: [1] Ferrocene is the first sandwitch compound discovered accidently in 1951. [2] Ferrocene is a diamagnetic yellow crystalline solid in which Fe is exist as zero oxidation state. [3] The melting point of ferrocene is 174 0C and boiling point is 249 0C.