Using the Linux Virtual Machine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Geany Tutorial
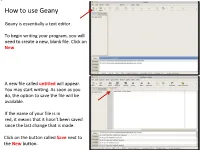
How to use Geany Geany is essentially a text editor. To begin writing your program, you will need to create a new, blank file. Click on New. A new file called untitled will appear. You may start writing. As soon as you do, the option to save the file will be available. If the name of your file is in red, it means that it hasn’t been saved since the last change that is made. Click on the button called Save next to the New button. Save the file in a directory you had previously created before you launched Geany and name it main.cpp. All of the files you will write and submit to will be named specifically main.cpp. Once the .cpp has been specified, Geany will turn on its color coding feature for the C++ template. Next, we will set up our environment and then write a simple program that will print something to the screen Feel free to supply your own name in this small program Before we do anything with it, we will need to configure some options to make your life easier in this class The vertical line to the right marks the ! boundary of your code. You will need to respect this limit in that any line of code you write must not cross this line and therefore be properly, manually broken down to the next line. Your code will be printed out for The line is not where it should be, however, and grading, and if your code crosses the we will now correct it line, it will cause line-wrapping and some points will be deducted. -

Software Tools: a Building Block Approach
SOFTWARE TOOLS: A BUILDING BLOCK APPROACH NBS Special Publication 500-14 U.S. DEPARTMENT OF COMMERCE National Bureau of Standards ] NATIONAL BUREAU OF STANDARDS The National Bureau of Standards^ was established by an act of Congress March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to pro- mote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Institute for Computer Sciences and Technology, the Office for Information Programs, and the ! Office of Experimental Technology Incentives Program. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consist- ent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essen- tial services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. The Institute consists of the Office of Measurement Services, and the following center and divisions: Applied Mathematics — Electricity — Mechanics — Heat — Optical Physics — Center for Radiation Research — Lab- oratory Astrophysics^ — Cryogenics^ — Electromagnetics^ — Time and Frequency*. THE INSTITUTE FOR MATERIALS RESEARCH conducts materials research leading to improved methods of measure- ment, standards, and data on the properties of well-characterized materials needed by industry, commerce, educational insti- tutions, and Government; provides advisory and research services to other Government agencies; and develops, produces, and distributes standard reference materials. -

Text Editing in UNIX: an Introduction to Vi and Editing
Text Editing in UNIX A short introduction to vi, pico, and gedit Copyright 20062009 Stewart Weiss About UNIX editors There are two types of text editors in UNIX: those that run in terminal windows, called text mode editors, and those that are graphical, with menus and mouse pointers. The latter require a windowing system, usually X Windows, to run. If you are remotely logged into UNIX, say through SSH, then you should use a text mode editor. It is possible to use a graphical editor, but it will be much slower to use. I will explain more about that later. 2 CSci 132 Practical UNIX with Perl Text mode editors The three text mode editors of choice in UNIX are vi, emacs, and pico (really nano, to be explained later.) vi is the original editor; it is very fast, easy to use, and available on virtually every UNIX system. The vi commands are the same as those of the sed filter as well as several other common UNIX tools. emacs is a very powerful editor, but it takes more effort to learn how to use it. pico is the easiest editor to learn, and the least powerful. pico was part of the Pine email client; nano is a clone of pico. 3 CSci 132 Practical UNIX with Perl What these slides contain These slides concentrate on vi because it is very fast and always available. Although the set of commands is very cryptic, by learning a small subset of the commands, you can edit text very quickly. What follows is an outline of the basic concepts that define vi. -

Basic Guide: Using VIM Introduction: VI and VIM Are In-Terminal Text Editors That Come Standard with Pretty Much Every UNIX Op
Basic Guide: Using VIM Introduction: VI and VIM are in-terminal text editors that come standard with pretty much every UNIX operating system. Our Astro computers have both. These editors are an alternative to using Emacs for editing and writing programs in python. VIM is essentially an extended version of VI, with more features, so for the remainder of this discussion I will be simply referring to VIM. (But if you ever do research and need to ssh onto another campus’s servers and they only have VI, 99% of what you know will still apply). There are advantages and disadvantages to using VIM, just like with any text editors. The main disadvantage of VIM is that it has a very steep learning curve, because it is operated via many keyboard shortcuts with somewhat obscure names like dd, dw, d}, p, u, :q!, etc. In addition, although VIM will do syntax highlighting for Python (ie, color code things based on what type of thing they are), it doesn’t have much intuitive support for writing long, extensive, and complex codes (though if you got comfortable enough you could conceivably do it). On the other hand, the advantage of VIM is once you learn how to use it, it is one of the most efficient ways of editing text. (Because of all the shortcuts, and efficient ways of opening and closing). It is perfectly reasonable to use a dedicated program like emacs, sublime, canopy, etc., for creating your code, and learning VIM as a way to edit your code on the fly as you try to run it. -

The Kate Handbook
The Kate Handbook Anders Lund Seth Rothberg Dominik Haumann T.C. Hollingsworth The Kate Handbook 2 Contents 1 Introduction 10 2 The Fundamentals 11 2.1 Starting Kate . 11 2.1.1 From the Menu . 11 2.1.2 From the Command Line . 11 2.1.2.1 Command Line Options . 12 2.1.3 Drag and Drop . 13 2.2 Working with Kate . 13 2.2.1 Quick Start . 13 2.2.2 Shortcuts . 13 2.3 Working With the KateMDI . 14 2.3.1 Overview . 14 2.3.1.1 The Main Window . 14 2.3.2 The Editor area . 14 2.4 Using Sessions . 15 2.5 Getting Help . 15 2.5.1 With Kate . 15 2.5.2 With Your Text Files . 16 2.5.3 Articles on Kate . 16 3 Working with the Kate Editor 17 4 Working with Plugins 18 4.1 Kate Application Plugins . 18 4.2 External Tools . 19 4.2.1 Configuring External Tools . 19 4.2.2 Variable Expansion . 20 4.2.3 List of Default Tools . 22 4.3 Backtrace Browser Plugin . 25 4.3.1 Using the Backtrace Browser Plugin . 25 4.3.2 Configuration . 26 4.4 Build Plugin . 26 The Kate Handbook 4.4.1 Introduction . 26 4.4.2 Using the Build Plugin . 26 4.4.2.1 Target Settings tab . 27 4.4.2.2 Output tab . 28 4.4.3 Menu Structure . 28 4.4.4 Thanks and Acknowledgments . 28 4.5 Close Except/Like Plugin . 28 4.5.1 Introduction . 28 4.5.2 Using the Close Except/Like Plugin . -

ENCM 335 Fall 2018: Command-Line C Programming on Macos
page 1 of 4 ENCM 335 Fall 2018: Command-line C programming on macOS Steve Norman Department of Electrical & Computer Engineering University of Calgary September 2018 Introduction This document is intended to help students who would like to do ENCM 335 C pro- gramming on an Apple Mac laptop or desktop computer. A note about software versions The information in this document was prepared using the latest versions of macOS Sierra version 10.12.6 and Xcode 9.2, the latest version of Xcode available for that version of macOS. Things should work for you if you have other but fairly recent versions of macOS and Xcode. If you have versions that are several years old, though you may experience difficulties that I'm not able to help with. Essential Software There are several key applications needed to do command-line C programming on a Mac. Web browser I assume you all know how to use a web browser to find course documents, so I won't say anything more about web browsers here. Finder Finder is the tool built in to macOS for browsing folders and files. It really helps if you're fluent with its features. Terminal You'll need Terminal to enter commands and look at output of commands. To start it up, look in Utilities, which is a folder within the Applications folder. It probably makes sense to drag the icon for Terminal to the macOS Dock, so that you can launch it quickly. macOS Terminal runs the same bash shell that runs in Cygwin Terminal, so commands like cd, ls, mkdir, and so on, are all available on your Mac. -

O'reilly GNU Emacs Pocket Reference.Pdf
Page i GNU Emacs Pocket Reference Debra Cameron Beijing • Cambridge • Farnham • Köln • Paris • Sebastopol • Taipei • Tokyo Page ii GNU Emacs Pocket Reference by Debra Cameron Copyright ã 1999 O'Reilly & Associates, Inc. All rights reserved. Printed in the United States of America. Editor: Gigi Estabrook Production Editor: Claire Cloutier LeBlanc Production Services: Omegatype Typography, Inc. Cover Design: Edie Freedman Printing History: January 1999: First Edition Nutshell Handbook, the Nutshell Handbook logo, and the O'Reilly logo are registered trademarks of O'Reilly & Associates, Inc. The association between the image of a gnu and the topic of GNU Emacs is a trademark of O'Reilly & Associates, Inc. Many of the designations used by manufacturers and sellers to distinguish their products are claimed as trademarks. Where those designations appear in this book, and O'Reilly & Associates, Inc. was aware of a trademark claim, the designations have been printed in caps or initial caps. While every precaution has been taken in the preparation of this book, the publisher assumes no responsibility for errors or omissions, or for damages resulting from the use of the information contained herein. This book is printed on acid-free paper with 85% recycled content, 15% post-consumer waste. O'Reilly & Associates is committed to using paper with the highest recycled content available consistent with high quality. ISBN: 1-56592-496-7 [11/99] Page iii Table of Contents Introduction 1 Emacs Commands 1 Conventions 2 1. Emacs Basics 2 2. Editing Files 5 3. Search and Replace Operations 10 4. Using Buffers and Windows 15 5. Emacs as a Work Environment 19 6. -

Command Line Interface (Shell)
Command Line Interface (Shell) 1 Organization of a computer system users applications graphical user shell interface (GUI) operating system hardware (or software acting like hardware: “virtual machine”) 2 Organization of a computer system Easier to use; users applications Not so easy to program with, interactive actions automate (click, drag, tap, …) graphical user shell interface (GUI) system calls operating system hardware (or software acting like hardware: “virtual machine”) 3 Organization of a computer system Easier to program users applications with and automate; Not so convenient to use (maybe) typed commands graphical user shell interface (GUI) system calls operating system hardware (or software acting like hardware: “virtual machine”) 4 Organization of a computer system users applications this class graphical user shell interface (GUI) operating system hardware (or software acting like hardware: “virtual machine”) 5 What is a Command Line Interface? • Interface: Means it is a way to interact with the Operating System. 6 What is a Command Line Interface? • Interface: Means it is a way to interact with the Operating System. • Command Line: Means you interact with it through typing commands at the keyboard. 7 What is a Command Line Interface? • Interface: Means it is a way to interact with the Operating System. • Command Line: Means you interact with it through typing commands at the keyboard. So a Command Line Interface (or a shell) is a program that lets you interact with the Operating System via the keyboard. 8 Why Use a Command Line Interface? A. In the old days, there was no choice 9 Why Use a Command Line Interface? A. -

SAS on Unix/Linux- from the Terminal to GUI
SAS on Unix/Linux- from the terminal to GUI. L Gakava & S Kannan – October 2015 Agenda All about the terminal o Customising your terminal o Basic Linux terminal commands o Running SAS in non-interactive mode o Available SAS file editors o What to look out for on Unix/Linux platform All about Graphical User Interface (GUI) o Launching SAS GUI. o Changing SAS default behaviour o SAS ToolBox commands o SAS editor commands. Motivation - Why Use SAS On Unix/Linux? Using SAS on UNIX/Linux Platform o Company migrating to UNIX/Linux o Joining a company which is using SAS on the Linux platform Challenge Too many commands to learn! Why Use SAS On Unix/Linux o Customising Linux sessions will ensure you increase work efficiency by taking advantage of the imbedded Linux tools. In general transferring and running large files will be quicker in Linux compared to PC*. Terminal What to expect when you login? % pwd /home/username % ls Customise: Update .bashrc file with this line PS1='$IV $PWD$EE> ' will change your prompt to show the following: /home/username> Terminal Navigation Command Meaning ls list files and directories ls -a list all files and directories mkdir make a directory cd directory change to named directory cd change to home-directory cd ~ change to home-directory cd .. change to parent directory Terminal Navigation Command Meaning cp file1 file2 copy file1 and call it file2 mv file1 file2 move or rename file1 to file2 rm file remove a file rmdir directory remove a directory cat file display a file less file display a file a page at a time head file display the first few lines of a file tail file display the last few lines of a file grep 'keyword' file search a file for keywords count number of lines/words/ wc file characters in file Terminal useful commands How do you find out if a version of a file has changed? /home/username>diff file1.txt file2.txt Command to compare two files. -

Geanypy Documentation Release 1.0
GeanyPy Documentation Release 1.0 Matthew Brush <[email protected]> February 17, 2017 Contents 1 Introduction 3 2 Installation 5 2.1 Getting the Source............................................5 2.2 Dependencies and where to get them..................................5 2.3 And finally ... installing GeanyPy....................................7 3 Getting Started 9 3.1 What the heck is GeanyPy, really?....................................9 3.2 Python Console..............................................9 3.3 Future Plans............................................... 10 4 Writing a Plugin - Quick Start Guide 11 4.1 The Plugin Interface........................................... 11 4.2 Real-world Example........................................... 12 4.3 Logging.................................................. 13 5 API Documentation 15 5.1 The app module............................................. 15 5.2 The dialogs module.......................................... 16 5.3 The document module......................................... 17 5.4 The geany package and module.................................... 19 6 Indices and tables 21 Python Module Index 23 i ii GeanyPy Documentation, Release 1.0 Contents: Contents 1 GeanyPy Documentation, Release 1.0 2 Contents CHAPTER 1 Introduction GeanyPy allows people to write their Geany plugins in Python making authoring a plugin much more accessible to non C programmers. What follows is a description of installing and using the GeanyPy plugin, paving the way for the rest of the documentation to covert the details of programming with the GeanyPy bindings of the Geany API. 3 GeanyPy Documentation, Release 1.0 4 Chapter 1. Introduction CHAPTER 2 Installation Currently there are no binary packages available for installing GeanyPy so it must be installed from source. The following instructions will describe how to do this. Getting the Source The best way currently to get GeanyPy is to check it out from it’s repository on GitHub.com. -

Programmable Command Languages for Window Systems Egie
Programmable Command Languages for Window Systems Rlchard Joel Cohn CMU-CS-88-139 June 1988 _egie Programmable Command Languages for Window Systems Report number CMU-CS-88-139 Copyright © 1988 by Richard Joel Cohn. All rights reserved. This dissertation was submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Computer Science at Carnegie Mellon University. The research in this dissertation was supported by the Information Technology Center, a joint project of Carnegie Mellon University and International Business Machines Corporation. This dissertation was produced with Scribe. Illustrations were produced with Pic, Troff, and TranScript. Aldus and PageMaker are trademarks of Aldus Corporation. Apple and Macintosh are registered trademarks of Apple Computer, Inc. Clads is a trademark of Clads Corporation. DEC is a trademark of Digital Equipment Corporation. Hypercard is a trademark of Apple Computer, Inc. IBM is a registered trademark of International Business Machines Corporation. MacDraw, MacPaint, and MacWrite are registered trademarks of Clads Corporation. NeWS and Sun-3 are trademarks of Sun Microsystems, Inc. PostScript and TranScript are trademarks of Adobe Systems, Inc. Scribe is a registered trademark of Scribe Systems. SmaUtalk-80is a trademark of Xerox Corporation. Tempo is a trademark of Affinity Microsystems, Ltd. UNIXis a registered trademark of AT&T Bell Laboratories. Abstract Programmable command and macro languages have long served as important tools for users of computer systems. This thesis describes the design and implementation of a general-purpose command language for a window system. This has never been tried be- fore because these systems are new and because the problems inherent in providing a command language for a window system are hard. -

System Programming, Homework Assignment 1 Due Wednesday, January 20
System Programming, Homework Assignment 1 Due Wednesday, January 20 Playing around on Earth For this assignment, you will get a chance to get used to using the command-line interface offered by the shell and using your chosen text editor. Even if you have a Unix machine of your own, you will need to complete this assignment on one of the department's general-purpose Unix systems (e.g., earth). Notice that I'm not asking you to turn in anything for this work. I'll take a look in your account on Wednesday to see if you've done these tasks. Also, at the start of class on Wednesday, we'll have a short quiz to see how much you've learned about your text editor. Log on to the Unix systems using putty, or some other secure shell client. If this is your first time using these systems, change your password using the passwd command. When your account was created, it was assigned a randomly-generated password. You will want to change this to something that's easy for you to remember but hard for anyone else to guess. Just type the passwd command at the shell prompt. This will prompt you for your old password and give you a chance to choose a new password. This change will automatically alter your password on all of our Unix machines. There is a similar command, chsh, that will change your default shell command. A number of alternative shells are available for people who prefer a slightly different command-line interface.