Downloaded from the Gtex Portal Ones Can Give Rise to Non-Specific Effects and Has Therapeutic (Version 7)
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
![Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]](https://docslib.b-cdn.net/cover/8283/computational-genome-wide-identification-of-heat-shock-protein-genes-in-the-bovine-genome-version-1-peer-review-2-approved-1-approved-with-reservations-88283.webp)
Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]
F1000Research 2018, 7:1504 Last updated: 08 AUG 2021 RESEARCH ARTICLE Computational genome-wide identification of heat shock protein genes in the bovine genome [version 1; peer review: 2 approved, 1 approved with reservations] Oyeyemi O. Ajayi1,2, Sunday O. Peters3, Marcos De Donato2,4, Sunday O. Sowande5, Fidalis D.N. Mujibi6, Olanrewaju B. Morenikeji2,7, Bolaji N. Thomas 8, Matthew A. Adeleke 9, Ikhide G. Imumorin2,10,11 1Department of Animal Breeding and Genetics, Federal University of Agriculture, Abeokuta, Nigeria 2International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, 14853, USA 3Department of Animal Science, Berry College, Mount Berry, GA, 30149, USA 4Departamento Regional de Bioingenierias, Tecnologico de Monterrey, Escuela de Ingenieria y Ciencias, Queretaro, Mexico 5Department of Animal Production and Health, Federal University of Agriculture, Abeokuta, Nigeria 6Usomi Limited, Nairobi, Kenya 7Department of Animal Production and Health, Federal University of Technology, Akure, Nigeria 8Department of Biomedical Sciences, Rochester Institute of Technology, Rochester, NY, 14623, USA 9School of Life Sciences, University of KwaZulu-Natal, Durban, 4000, South Africa 10School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, 30032, USA 11African Institute of Bioscience Research and Training, Ibadan, Nigeria v1 First published: 20 Sep 2018, 7:1504 Open Peer Review https://doi.org/10.12688/f1000research.16058.1 Latest published: 20 Sep 2018, 7:1504 https://doi.org/10.12688/f1000research.16058.1 Reviewer Status Invited Reviewers Abstract Background: Heat shock proteins (HSPs) are molecular chaperones 1 2 3 known to bind and sequester client proteins under stress. Methods: To identify and better understand some of these proteins, version 1 we carried out a computational genome-wide survey of the bovine 20 Sep 2018 report report report genome. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle
Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle Slim Ben-Jemaa, Gabriele Senczuk, Elena Ciani, Roberta Ciampolini, Gennaro Catillo, Mekki Boussaha, Fabio Pilla, Baldassare Portolano, Salvatore Mastrangelo To cite this version: Slim Ben-Jemaa, Gabriele Senczuk, Elena Ciani, Roberta Ciampolini, Gennaro Catillo, et al.. Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle. Frontiers in Genetics, Frontiers, 2021, 10.3389/fgene.2021.675569. hal-03210766 HAL Id: hal-03210766 https://hal.inrae.fr/hal-03210766 Submitted on 28 Apr 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ORIGINAL RESEARCH published: 28 April 2021 doi: 10.3389/fgene.2021.675569 Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle Slim Ben-Jemaa 1, Gabriele Senczuk 2, Elena Ciani 3, Roberta -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Location Analysis of Estrogen Receptor Target Promoters Reveals That
Location analysis of estrogen receptor ␣ target promoters reveals that FOXA1 defines a domain of the estrogen response Jose´ e Laganie` re*†, Genevie` ve Deblois*, Ce´ line Lefebvre*, Alain R. Bataille‡, Franc¸ois Robert‡, and Vincent Gigue` re*†§ *Molecular Oncology Group, Departments of Medicine and Oncology, McGill University Health Centre, Montreal, QC, Canada H3A 1A1; †Department of Biochemistry, McGill University, Montreal, QC, Canada H3G 1Y6; and ‡Laboratory of Chromatin and Genomic Expression, Institut de Recherches Cliniques de Montre´al, Montreal, QC, Canada H2W 1R7 Communicated by Ronald M. Evans, The Salk Institute for Biological Studies, La Jolla, CA, July 1, 2005 (received for review June 3, 2005) Nuclear receptors can activate diverse biological pathways within general absence of large scale functional data linking these putative a target cell in response to their cognate ligands, but how this binding sites with gene expression in specific cell types. compartmentalization is achieved at the level of gene regulation is Recently, chromatin immunoprecipitation (ChIP) has been used poorly understood. We used a genome-wide analysis of promoter in combination with promoter or genomic DNA microarrays to occupancy by the estrogen receptor ␣ (ER␣) in MCF-7 cells to identify loci recognized by transcription factors in a genome-wide investigate the molecular mechanisms underlying the action of manner in mammalian cells (20–24). This technology, termed 17-estradiol (E2) in controlling the growth of breast cancer cells. ChIP-on-chip or location analysis, can therefore be used to deter- We identified 153 promoters bound by ER␣ in the presence of E2. mine the global gene expression program that characterize the Motif-finding algorithms demonstrated that the estrogen re- action of a nuclear receptor in response to its natural ligand. -

Protein Identities in Evs Isolated from U87-MG GBM Cells As Determined by NG LC-MS/MS
Protein identities in EVs isolated from U87-MG GBM cells as determined by NG LC-MS/MS. No. Accession Description Σ Coverage Σ# Proteins Σ# Unique Peptides Σ# Peptides Σ# PSMs # AAs MW [kDa] calc. pI 1 A8MS94 Putative golgin subfamily A member 2-like protein 5 OS=Homo sapiens PE=5 SV=2 - [GG2L5_HUMAN] 100 1 1 7 88 110 12,03704523 5,681152344 2 P60660 Myosin light polypeptide 6 OS=Homo sapiens GN=MYL6 PE=1 SV=2 - [MYL6_HUMAN] 100 3 5 17 173 151 16,91913397 4,652832031 3 Q6ZYL4 General transcription factor IIH subunit 5 OS=Homo sapiens GN=GTF2H5 PE=1 SV=1 - [TF2H5_HUMAN] 98,59 1 1 4 13 71 8,048185945 4,652832031 4 P60709 Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 - [ACTB_HUMAN] 97,6 5 5 35 917 375 41,70973209 5,478027344 5 P13489 Ribonuclease inhibitor OS=Homo sapiens GN=RNH1 PE=1 SV=2 - [RINI_HUMAN] 96,75 1 12 37 173 461 49,94108966 4,817871094 6 P09382 Galectin-1 OS=Homo sapiens GN=LGALS1 PE=1 SV=2 - [LEG1_HUMAN] 96,3 1 7 14 283 135 14,70620005 5,503417969 7 P60174 Triosephosphate isomerase OS=Homo sapiens GN=TPI1 PE=1 SV=3 - [TPIS_HUMAN] 95,1 3 16 25 375 286 30,77169764 5,922363281 8 P04406 Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 - [G3P_HUMAN] 94,63 2 13 31 509 335 36,03039959 8,455566406 9 Q15185 Prostaglandin E synthase 3 OS=Homo sapiens GN=PTGES3 PE=1 SV=1 - [TEBP_HUMAN] 93,13 1 5 12 74 160 18,68541938 4,538574219 10 P09417 Dihydropteridine reductase OS=Homo sapiens GN=QDPR PE=1 SV=2 - [DHPR_HUMAN] 93,03 1 1 17 69 244 25,77302971 7,371582031 11 P01911 HLA class II histocompatibility antigen, -

Sequence Composition and Evolution of Mammalian B Chromosomes
G C A T T A C G G C A T genes Review Sequence Composition and Evolution of Mammalian B Chromosomes Nikolay B. Rubtsov 1,2,* and Yury M. Borisov 3 1 The Federal Research Center Institute of Cytology and Genetics SB RAS, Lavrentiev Ave. 10, Novosibirsk 630090, Russia 2 Novosibirsk State University, Pirogova Str. 2, Novosibirsk 630090, Russia 3 Severtzov Institute of Ecology and Evolution, Russia Academy of Sciences, Leninsky Pr. 33, Moscow 119071, Russia; [email protected] * Correspondence: [email protected]; Tel.: +7-913-941-5682 Received: 28 August 2018; Accepted: 1 October 2018; Published: 10 October 2018 Abstract: B chromosomes (Bs) revealed more than a hundred years ago remain to be some of the most mysterious elements of the eukaryotic genome. Their origin and evolution, DNA composition, transcriptional activity, impact on adaptiveness, behavior in meiosis, and transfer to the next generation require intensive investigations using modern methods. Over the past years, new experimental techniques have been applied and helped us gain a deeper insight into the nature of Bs. Here, we consider mammalian Bs, taking into account data on their DNA sequencing, transcriptional activity, positions in nuclei of somatic and meiotic cells, and impact on genome functioning. Comparative cytogenetics of Bs suggests the existence of different mechanisms of their formation and evolution. Due to the long and complicated evolvement of Bs, the similarity of their morphology could be explained by the similar mechanisms involved in their development while the difference between Bs even of the same origin could appear due to their positioning at different stages of their evolution. -

Global Analysis of Chaperone Effects Using a Reconstituted Cell-Free Translation System
Global analysis of chaperone effects using a reconstituted cell-free translation system Tatsuya Niwaa, Takashi Kanamorib,1, Takuya Uedab,2, and Hideki Taguchia,2 aDepartment of Biomolecular Engineering, Graduate School of Biosciences and Biotechnology, Tokyo Institute of Technology, Midori-ku, Yokohama 226-8501, Japan; and bDepartment of Medical Genome Sciences, Graduate School of Frontier Sciences, University of Tokyo, Kashiwa, Chiba 277-8562, Japan Edited by George H. Lorimer, University of Maryland, College Park, MD, and approved April 19, 2012 (received for review January 25, 2012) Protein folding is often hampered by protein aggregation, which can three chaperone systems are known to act cooperatively: TF and be prevented by a variety of chaperones in the cell. A dataset that DnaK exhibit overlapping cotranslational roles in vivo (13–15). evaluates which chaperones are effective for aggregation-prone Overexpression of DnaK/DnaJ and GroEL/GroES in E. coli proteins would provide an invaluable resource not only for un- rpoH mutant cells, which are deficient in heat-shock proteins, derstanding the roles of chaperones, but also for broader applications prevents aggregation of newly translated proteins (16). GroEL is in protein science and engineering. Therefore, we comprehensively believed to be involved in folding after the polypeptides are re- evaluated the effects of the major Escherichia coli chaperones, trigger leased from the ribosome, although the possible cotranslational factor, DnaK/DnaJ/GrpE, and GroEL/GroES, on ∼800 aggregation- involvement of GroEL has also been reported (17–20). prone cytosolic E. coli proteins, using a reconstituted chaperone-free Over the past two decades many efforts have been focused on translation system. -
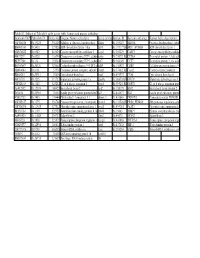
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

Anti-HSPA14 Antibody (ARG41869)
Product datasheet [email protected] ARG41869 Package: 100 μl anti-HSPA14 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes HSPA14 Tested Reactivity Hu, Ms, Mk Tested Application WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name HSPA14 Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 240-509 of Human HSPA14. (NP_057383.2) Conjugation Un-conjugated Alternate Names HSP70-4; HSP70-like protein 1; Heat shock protein HSP60; Heat shock 70 kDa protein 14; HSP70L1 Application Instructions Application table Application Dilution WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control HeLa Calculated Mw 55 kDa Observed Size ~ 57 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol HSPA14 Gene Full Name heat shock 70kDa protein 14 Function Component of the ribosome-associated complex (RAC), a complex involved in folding or maintaining nascent polypeptides in a folding-competent state. In the RAC complex, binds to the nascent polypeptide chain, while DNAJC2 stimulates its ATPase activity. -

Α Are Regulated by Heat Shock Protein 90
The Levels of Retinoic Acid-Inducible Gene I Are Regulated by Heat Shock Protein 90- α Tomoh Matsumiya, Tadaatsu Imaizumi, Hidemi Yoshida, Kei Satoh, Matthew K. Topham and Diana M. Stafforini This information is current as of October 2, 2021. J Immunol 2009; 182:2717-2725; ; doi: 10.4049/jimmunol.0802933 http://www.jimmunol.org/content/182/5/2717 Downloaded from References This article cites 44 articles, 19 of which you can access for free at: http://www.jimmunol.org/content/182/5/2717.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology The Levels of Retinoic Acid-Inducible Gene I Are Regulated by Heat Shock Protein 90-␣1 Tomoh Matsumiya,*‡ Tadaatsu Imaizumi,‡ Hidemi Yoshida,‡ Kei Satoh,‡ Matthew K. Topham,*† and Diana M. Stafforini2*† Retinoic acid-inducible gene I (RIG-I) is an intracellular pattern recognition receptor that plays important roles during innate immune responses to viral dsRNAs.