The Total Synthesis of Natural Products Possessing Biological Activity in Two Important Areas
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Porphyrin Carbene Complexes: (5,10,15,20-Tetra-P-Tolylporphyrinato )
Chemistry Publications Chemistry 8-1994 Properties and Molecular Structures of Osmium(ll) Porphyrin Carbene Complexes: (5,10,15,20-Tetra-p-tolylporphyrinato )osmium Di-p-tolylmethylidene and (5,10,15,20-Tetra-p- tolylporphyrinato)osmium (Trimethylsilyl)methylidene Jean-Pierre Djukic Iowa State University Daniel A. Smith Iowa State University Victor G. Young Jr. Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/chem_pubs L. Keith Woo IowaP Satrate of U ntheiversitCyhe, kmiwoo@istryas Ctaommonte.edu s The ompc lete bibliographic information for this item can be found at http://lib.dr.iastate.edu/ chem_pubs/727. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Chemistry at Iowa State University Digital Repository. It has been accepted for inclusion in Chemistry Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Properties and Molecular Structures of Osmium(ll) Porphyrin Carbene Complexes: (5,10,15,20-Tetra-p-tolylporphyrinato )osmium Di-p- tolylmethylidene and (5,10,15,20-Tetra-p-tolylporphyrinato)osmium (Trimethylsilyl)methylidene Abstract The first molecular structures of two (porphyrinato)osmium(II) alkylidene complexes are described. The carbene fragments of (5,10,15,20-tetra-p-tolylporphyrinato)osmium (trimethylsilyl) methylidene (1) and (5,10,15,20-tetra-p-tolylporphyrinato)osmium di-p-tolylmethylidene (2) adopt different conformations in the solid state. With respect to the porphyrin ring nitrogen atoms, a staggered conformation is found for the complex 1 carbene moiety (dos-e = 1. -

Optimizing the Least Nucleophilic Anion. a New, Strong Methyl+ Reagent Daniel Stasko and Christopher A
Published on Web 01/25/2002 Optimizing the Least Nucleophilic Anion. A New, Strong Methyl+ Reagent Daniel Stasko and Christopher A. Reed* Department of Chemistry, UniVersity of California, RiVerside, California 92521-0304 Received August 3, 2001 The ideal weakly coordinating counterion for reactive cations would be the least nucleophilic, least basic, most inert anion available. It should also be inexpensive, have useful spectroscopic handles, and confer good solubility and crystallizing properties on - its salts. Triflate anion, CF3SO3 , has served chemistry well in this regard but larger size, absence of lone pairs, and extreme chemical inertness have recently made carboranes1 or fluorinated tetraphe- nylborates the preferred choice for many applications.2 These anions have led to the solution of the silylium ion problem,3 enhanced Lewis acid catalysis by Li+ ion,4-6 new “strong-but-gentle” superacids,7 commercially viable olefin polymerization catalysts,8 new possibilities for electrolytes9,10 and ionic liquids,11 and the •+ 7 + 12 isolation of new reactive cations such as C60 , Bu3Sn , Cu- + 13 (CO)4 , etc. Figure 1. The 1-H-2,3,4,5,6-pentamethyl-7,8,9,10,11,12-hexahalo-CB11 - ) The perfluorinated tetraphenylborate anion is the preferred choice carborane anions, 1-H-CB11Me5X6 (X Cl, Br, I), used in this work. for price and solubility reasons but its salts frequently form liquid 29 Table 1. Selected Si Chemical Shifts (ppm) for i-Pr3SiY clathrates and fail to crystallize. The boron-phenyl bond is rather 29 easily cleaved by strong Brønsted14 or Lewis15 acids. Halogenated compd Si conditions carborane anions are much more inert than tetraphenylborates, their i-Pr3Si(1-H-CB11H5Br6) 100 C6D6 salts crystallize well, but they are more expensive and their salts i-Pr3Si(1-H-CB11H5Cl6) 103 C6D6 i-Pr Si(F -BPh )a 108 solid state less soluble. -
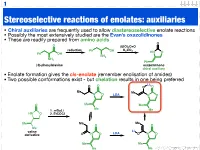
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Cyanosilylation of Aldehydes Catalyzed by Ag(I)- and Cu(II)-Arylhydrazone Coordination Polymers in Conventional and in Ionic Liquid Media
catalysts Article Cyanosilylation of Aldehydes Catalyzed by Ag(I)- and Cu(II)-Arylhydrazone Coordination Polymers in Conventional and in Ionic Liquid Media Gonçalo A. O. Tiago 1, Kamran T. Mahmudov 1,2,*, M. Fátima C. Guedes da Silva 1,* , Ana P. C. Ribeiro 1,* , Luís C. Branco 3, Fedor I. Zubkov 4 and Armando J. L. Pombeiro 1 1 Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049–001 Lisboa, Portugal; [email protected] (G.A.O.T.); [email protected] (A.J.L.P.) 2 Department of Chemistry, Baku State University, Z. Xalilov Str. 23, Az 1148 Baku, Azerbaijan 3 LAQV-REQUINTE, Departamento de Química, Faculdade de Ciências e Tecnologias da Universidade Nova de Lisboa, Quinta da Torre, 2829-516 Caparica, Portugal; [email protected] 4 Organic Chemistry Department, Faculty of Science, Peoples’ Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., Moscow 117198, Russian; [email protected] * Correspondence: [email protected] or [email protected] (K.T.M.); [email protected] (M.F.C.G.d.S.); [email protected] (A.P.C.R.) Received: 22 February 2019; Accepted: 15 March 2019; Published: 20 March 2019 0 Abstract: The novel Ag(I) and Cu(II) coordination polymers [Ag(m3-1κO;2:3κO ;4κN-HL)]n·n/2H2O(1) − and [Cu(en)2(m-1κO;2κN-L)]n·nH2O(2) [HL = 2-(2-(1-cyano-2-oxopropylidene)hydrazinyl)benzene sulfonate] were synthesized and characterized by IR and ESI-MS spectroscopies, elemental and single crystal X-ray diffraction analyses. -

Trimethylsilyl Trifluoromethanesulfonate-Mediated Additions to Acetals, Nitrones, and Aminals Chelsea Safran
University of Richmond UR Scholarship Repository Honors Theses Student Research 4-1-2013 Trimethylsilyl trifluoromethanesulfonate-mediated additions to acetals, nitrones, and aminals Chelsea Safran Follow this and additional works at: http://scholarship.richmond.edu/honors-theses Recommended Citation Safran, Chelsea, "Trimethylsilyl trifluoromethanesulfonate-mediated additions to acetals, nitrones, and aminals" (2013). Honors Theses. Paper 71. This Thesis is brought to you for free and open access by the Student Research at UR Scholarship Repository. It has been accepted for inclusion in Honors Theses by an authorized administrator of UR Scholarship Repository. For more information, please contact [email protected]. Trimethylsilyl trifluoromethanesulfonate-mediated additions to acetals, nitrones, and aminals By Chelsea Safran Honors Thesis In Program In Biochemistry and Molecular Biology University of Richmond Richmond, VA Spring 2012 Advisor: Dr. C. Wade Downey This thesis has been accepted as part of the honors requirements in the Program in Biochemistry and Molecular Biology ______________________________ _________________ (advisor signature) (date) ______________________________ _________________ (reader signature) (date) Table of Contents i. Acknowledgements ii ii. Abstract iii iii. Chapter I: Introduction 1-4 iv. Chapter II: Amides 4-15 v. Chapter III: I. Bisthione Synthesis 16-18 II. Reactions with other N,O-acetals 18-22 vi. Chapter IV: I. Additions to Nitrones 22-25 II. Future Work 25 vii. Chapter V: Experimental I. N,O-acetal Formation 25-28 II. Addition to Nitrones 28-29 viii. Chapter VI: References 30 i Acknowledgments I would like to acknowledge my research Dr. Wade Downey for all of his time and dedication to my research for the past two years. -

IV (Advance Organic Synthesis and Supramolecular Chemistry and Carbocyclic Rings) Module No and 9: Protection and Deprotection Title Module Tag CHE P14 M9
Subject Chemistry Paper No and Title 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No and 9: Protection and deprotection Title Module Tag CHE_P14_M9 CHEMISTRY Paper No. 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No. 9: Protection and deprotection Table of Content 1. Learning outcomes 2. Introduction 3. Protecting groups for carbonyl compounds 4. Protecting groups for alcohols 5. Protecting groups for amines 6. Summary CHEMISTRY Paper No. 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No. 9: Protection and deprotection 1. Learning Outcomes After studying this module, you shall be able to Know about protecting groups. Study the characteristics of protecting groups. Understand the functional group protection. Know the protection of important functional groups. 2. Introduction Protection and deprotection is an important part of organic synthesis. During the course of synthesis, we many times desire to perform reaction at only one of the two functional groups in any single organic molecules. For example, in an organic compound possessing two functional groups like ester and ketone, we have to perform reaction at only ester group, them the keto group needs to be protected. If we want to reduce the ester group, then keto group will also get reduced. To avoid this type of complications, protection and deprotection of functional groups are necessary. 3. Protecting groups for carbonyl compounds The protecting groups allow the masking of a particular functional group where a specified reaction is not to be performed. The protection is required as it interferes with another reaction. -

Divergent Enantioselective Synthesis of Hapalindole-Type Alkaloids Using Catalytic Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Divergent enantioselective synthesis of hapalindole-type alkaloids using catalytic Cite this: Chem. Sci.,2016,7,4725 asymmetric hydrogenation of a ketone to construct the chiral core structure† Yang Liu,‡a Li-Jie Cheng,‡a Hai-Tao Yue,a Wen Che,a Jian-Hua Xie*a and Qi-Lin Zhouab A divergent enantioselective approach to hapalindole-type alkaloids is described. The route features a ruthenium-catalyzed asymmetric hydrogenation of a ketone via DKR to construct the chiral trans-1- indolyl-2-isopropenylcyclohexane skeleton and a switchable sequence of methylation and acetylation/ aldol reaction to access a chiral quaternary stereocenter. (+)-Hapalindole Q (1, 13 steps, 5.9% overall Received 15th February 2016 yield), (À)-12-epi-hapalindole Q isonitrile (2, 15 steps, 5.5% overall yield), (À)-hapalindole D (3, 14 steps, Accepted 12th April 2016 2.3% overall yield), and (+)-12-epi-fischerindole U isothiocyanate (4, 14 steps, 3.0% overall yield) were Creative Commons Attribution-NonCommercial 3.0 Unported Licence. DOI: 10.1039/c6sc00686h synthesized in 13–15 steps from a commercially available material to demonstrate the application of this www.rsc.org/chemicalscience approach. Introduction (+)-p-menth-1-en-9-ol.3,5f However, only one catalytic enantiose- lective synthesis of a hapalindole-type alkaloid has been re- 7 Owing to the unique and diverse molecular architectures of ported: Kinsman and Kerr used an organocatalyzed hapalindole-type alkaloids and their broad range of biological asymmetric Diels–Alder reaction as a key step in the synthesis of activities, they have recently attracted great interest as synthetic (+)-hapalindole Q (1). -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Aldol Reactions: E-Enolates and Anti-Selectivity
Utah State University DigitalCommons@USU All Graduate Plan B and other Reports Graduate Studies 5-2005 Aldol Reactions: E-Enolates and Anti-Selectivity Matthew Grant Anderson Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/gradreports Part of the Organic Chemistry Commons Recommended Citation Anderson, Matthew Grant, "Aldol Reactions: E-Enolates and Anti-Selectivity" (2005). All Graduate Plan B and other Reports. 1312. https://digitalcommons.usu.edu/gradreports/1312 This Report is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Plan B and other Reports by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. ALDOL REACTIONS: E-ENOLATES AND ANTI-SELECTIVITY Prepared By: MATTHEW GRANT ANDERSON A non-thesis paper submitted in partial fulfillment of the requirement for a Plan B Degree of Masters of Science in Organic Chemistry UTAH STATE UNIVERSITY Logan, Utah 2005 Contents Page CONTENTS ...................................................................................... .i LIST OF TABLES, FIGURES AND SCHEMES ....................................... ii,iii ABSTRACT .................................................................................... iv CHAPTER I. ALDOL REACTIONS:E-ENOLATES AND ANTI SELECTIVITY ......... 1 CHAPTER II. SECTION 1. MODELS OF E-ENOLATE FORMATION ...... .... ....... ... 12 SECTION 2. PATERSON ENOLATE PAPER ..... ......................... -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

Chapter 3. the Concept of Protecting Functional Groups
Chapter 3. The Concept of Protecting Functional Groups When a chemical reaction is to be carried out selectively at one reactive site in a multifunctional compound, other reactive sites must be temporarily blocked. A protecting group must fulfill a number of requirements: • The protecting group reagent must react selectively (kinetic chemoselectivity) in good yield to give a protected substrate that is stable to the projected reactions. • The protecting group must be selectively removed in good yield by readily available reagents. • The protecting group should not have additional functionality that might provide additional sites of reaction. 3.1 Protecting of NH groups Primary and secondary amines are prone to oxidation, and N-H bonds undergo metallation on exposure to organolithium and Grignard reagents. Moreover, the amino group possesses a lone pair electrons, which can be protonated or reacted with electrophiles. To render the lone pair electrons less reactive, the amine can be converted into an amide via acylation. N-Benzylamine Useful for exposure to organometallic reagents or metal hydrides Hydrogenolysis Benzylamines are not cleaved by Lewis acid Pearlman’s catalyst Amides Basicity of nitrogen is reduced, making them less susceptible to attack by electrophilic reagent The group is stable to pH 1-14, nucleophiles, organometallics (except organolithium reagents), catalytic hydrogenation, and oxidation. Cleaved by strong acid (6N HCl, HBr) or diisobutylaluminum hydride Carbamates Behave like a amides, hence no longer act as nucleophile Stable to oxidizing agents and aqueous bases but may react with reducing agents. Iodotrimethylsilane is often the reagent for removal of this protecting group Stable to both aqueous acid and base Benzoyloxycarbonyl group for peptide synthesis t-butoxycarbonyl group(Boc) is inert to hydrogenolysis and resistant to bases and nucleophilic reagent.