Chapter Template
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Folate Cycle Enzyme MTHFD1L Confers Metabolic Advantages in Hepatocellular Carcinoma
RESEARCH ARTICLE The Journal of Clinical Investigation Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma Derek Lee,1 Iris Ming-Jing Xu,1 David Kung-Chun Chiu,1 Robin Kit-Ho Lai,1 Aki Pui-Wah Tse,1 Lynna Lan Li,1 Cheuk-Ting Law,1 Felice Ho-Ching Tsang,1 Larry Lai Wei,1 Cerise Yuen-Ki Chan,1 Chun-Ming Wong,1,2 Irene Oi-Lin Ng,1,2 and Carmen Chak-Lui Wong1,2 1Department of Pathology and 2State Key Laboratory for Liver Research, The University of Hong Kong, Hong Kong, China. Cancer cells preferentially utilize glucose and glutamine, which provide macromolecules and antioxidants that sustain rapid cell division. Metabolic reprogramming in cancer drives an increased glycolytic rate that supports maximal production of these nutrients. The folate cycle, through transfer of a carbon unit between tetrahydrofolate and its derivatives in the cytoplasmic and mitochondrial compartments, produces other metabolites that are essential for cell growth, including nucleotides, methionine, and the antioxidant NADPH. Here, using hepatocellular carcinoma (HCC) as a cancer model, we have observed a reduction in growth rate upon withdrawal of folate. We found that an enzyme in the folate cycle, methylenetetrahydrofolate dehydrogenase 1–like (MTHFD1L), plays an essential role in support of cancer growth. We determined that MTHFD1L is transcriptionally activated by NRF2, a master regulator of redox homeostasis. Our observations further suggest that MTHFD1L contributes to the production and accumulation of NADPH to levels that are sufficient to combat oxidative stress in cancer cells. The elevation of oxidative stress through MTHFD1L knockdown or the use of methotrexate, an antifolate drug, sensitizes cancer cells to sorafenib, a targeted therapy for HCC. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Anti-MTHFD1 Monoclonal Antibody (DCABH- 12465) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-MTHFD1 monoclonal antibody (DCABH- 12465) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Antigen Description This gene encodes a protein that possesses three distinct enzymatic activities, 5,10- methylenetetrahydrofolate dehydrogenase, 5,10-methenyltetrahydrofolate cyclohydrolase and 10-formyltetrahydrofolate synthetase. Each of these activities catalyzes one of three sequential reactions in the interconversion of 1-carbon derivatives of tetrahydrofolate, which are substrates for methionine, thymidylate, and de novo purine syntheses. The trifunctional enzymatic activities are conferred by two major domains, an aminoterminal portion containing the dehydrogenase and cyclohydrolase activities and a larger synthetase domain. Immunogen A synthetic peptide of human MTHFD1 is used for rabbit immunization. Isotype IgG Source/Host Rabbit Species Reactivity Human Purification Protein A Conjugate Unconjugated Applications Western Blot (Transfected lysate); ELISA Size 1 ea Buffer In 1x PBS, pH 7.4 Preservative None Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. GENE INFORMATION Gene Name MTHFD1 methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1, methenyltetrahydrofolate cyclohydrolase, formyltetrahydrofolate synthetase [ Homo sapiens ] Official Symbol MTHFD1 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Synonyms MTHFD1; methylenetetrahydrofolate -
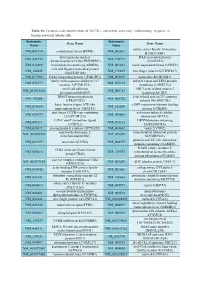
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

MTHFD1 Is a Genetic Interactor of BRD4 and Links Folate Metabolism to Transcriptional Regulation
bioRxiv preprint doi: https://doi.org/10.1101/439422; this version posted October 10, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. MTHFD1 is a genetic interactor of BRD4 and links folate metabolism to transcriptional regulation Sara Sdelci1, André F. Rendeiro1, Philipp Rathert2,3, Gerald Hofstätter1,4, Anna Ringler1,4, Herwig P. Moll5, Wanhui You1,4, Kristaps Klavins1, Bettina Gürtl1, Matthias Farlik1, Sandra Schick1,4, Freya Klepsch1, Matthew Oldach1, Pisanu Buphamalai1, Fiorella Schischlik1, Peter Májek1, Katja Parapatics1, Christian Schmidl1, Michael Schuster1, Thomas Penz1, Dennis L. Buckley6, Otto Hudecz7, Richard Imre7, Robert Kralovics1, Keiryn L. Bennett1, Andre C. Müller1, Karl Mechtler2, Jörg Menche1, James E. Bradner6, Georg E. Winter1, Emilio Casanova5,8, Christoph Bock1,9,10, Johannes Zuber2, Stefan Kubicek1,4,* Affiliations: 1CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences. Lazarettgasse 14, 1090 Vienna, Austria. 2Research Institute of Molecular Pathology (IMP), Vienna Biocenter (VBC), 1030 Vienna, Austria. 3Current address: Institute for Biochemistry (IBC), University Stuttgart, 70569 Stuttgart, Germany. 4Christian Doppler Laboratory for Chemical Epigenetics and Antiinfectives, CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 5Department of Physiology, Center of Physiology and Pharmacology & Comprehensive Cancer Center (CCC), Medical University of Vienna, Vienna, Austria 6Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA 02215 7Institute of Molecular Biotechnology, Austrian Academy of Sciences, Vienna Biocenter (VBC), 1030 Vienna, Austria. 8Ludwig Boltzmann Institute for Cancer Research (LBI-CR), Vienna, Austria 9Department of Laboratory Medicine, Medical University of Vienna, 1090 Vienna, Austria. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Differentially Expressed Genes Yuki
supplementary Table 1. The differentially expressed genes in F3-treated THP-1 cells after 6 hours. Gene Symbol Chip ID Fold Change Genbank UniGene Description CCL20 205476_at 421.7 NM_004591 Hs.75498 chemokine (C-C motif) ligand 20 CCL4 204103_at 351.6 NM_002984 Hs.75703 chemokine (C-C motif) ligand 4 BCL2A1 205681_at 228.6 NM_004049 Hs.227817 BCL2-related protein A1 ISG20 204698_at 202.1 NM_002201 Hs.459265 interferon stimulated exonuclease gene 20kDa CCL8 214038_at 139.1 AI984980 Hs.271387 chemokine (C-C motif) ligand 8 CXCL1 204470_at 115.1 NM_001511 Hs.789 chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating activity, alpha) IL7R 205798_at 111.6 NM_002185 Hs.362807 interleukin 7 receptor ; interleukin 7 receptor BCL3 204908_s_at 104.2 NM_005178 Hs.31210 B-cell CLL/lymphoma 3 GBP1 202269_x_at 99.99 BC002666 Hs.62661 guanylate binding protein 1, interferon-inducible, 67kDa SLAMF7 219159_s_at 98.3 NM_021181 Hs.517265 SLAM family member 7 CXCL10 204533_at 93.05 NM_001565 Hs.413924 chemokine (C-X-C motif) ligand 10 IL8 211506_s_at 91.31 AF043337 Hs.624 interleukin 8 IL8 202859_x_at 85.26 NM_000584 Hs.624 interleukin 8 THBS1 201110_s_at 84.5 NM_003246 Hs.164226 thrombospondin 1 EBI3 219424_at 82.87 NM_005755 Hs.501452 Epstein-Barr virus induced gene 3 SAT 213988_s_at 76.47 BE971383 Hs.28491 spermidine/spermine N1-acetyltransferase USP18 219211_at 76.22 NM_017414 Hs.38260 ubiquitin specific peptidase 18 LAMP3 205569_at 75.89 NM_014398 Hs.518448 lysosomal-associated membrane protein 3 CCL2 216598_s_at 71.33 S69738 Hs.303649 chemokine -

Downloaded from Consensuspathdb Collection We Defined High-Confidence MTHFD2-Interacting Pro- (
Koufaris and Nilsson Cancer & Metabolism (2018) 6:12 https://doi.org/10.1186/s40170-018-0185-4 RESEARCH Open Access Protein interaction and functional data indicate MTHFD2 involvement in RNA processing and translation Costas Koufaris1,2,3 and Roland Nilsson1,2,3* Abstract Background: The folate-coupled metabolic enzyme MTHFD2 is overexpressed in many tumor types and required for cancer cell proliferation, and is therefore of interest as a potential cancer therapeutic target. However, recent evidence suggests that MTHFD2 has a non-enzymatic function which may underlie the dependence of cancer cells on this protein. Understanding this non-enzymatic function is important for optimal targeting of MTHFD2 in cancer. Methods: To identify potential non-enzymatic functions of MTHFD2, we defined its interacting proteins using co-immunoprecipitation and mass spectrometry and integrated this information with large-scale co-expression analysis, protein dynamics, and gene expression response to MTHFD2 knockdown. Results: We found that MTHFD2 physically interacts with a set of nuclear proteins involved in RNA metabolism and translation, including components of the small ribosomal subunit and multiple members of the RNA-processing hnRNP family. Interacting proteins were also in general co-expressed with MTHFD2 in experiments that stimulate or repress proliferation, suggesting a close functional relationship. Also, unlike other folate one-carbon enzymes, the MTHFD2 protein has a short half-life and responds rapidly to serum. Finally, shRNA against MTHFD2 depletes several of its interactors and yields an overall transcriptional response similar to targeted inhibition of certain ribosomal subunits. Conclusions: Taken together, our findings suggest a novel function of MTHFD2 in RNA metabolism and translation. -

Mouse Mthfd2l Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Mthfd2l Knockout Project (CRISPR/Cas9) Objective: To create a Mthfd2l knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Mthfd2l gene (NCBI Reference Sequence: NM_026788 ; Ensembl: ENSMUSG00000029376 ) is located on Mouse chromosome 5. 8 exons are identified, with the ATG start codon in exon 1 and the TAG stop codon in exon 8 (Transcript: ENSMUST00000071652). Exon 2~3 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from about 11.54% of the coding region. Exon 2~3 covers 30.37% of the coding region. The size of effective KO region: ~2156 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 8 Legends Exon of mouse Mthfd2l Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 3 is aligned with itself to determine if there are tandem repeats. -

Methylenetetrahydrofolate Dehydrogenase 1 (MTHFD1) Is a the Human CCRCC Cell Line, Caki-1, in Vitro, and the Possible Hinge Enzyme in the Folic Acid Metabolic Pathway
LAB/IN VITRO RESEARCH e-ISSN 1643-3750 © Med Sci Monit, 2018; 24: 8391-8400 DOI: 10.12659/MSM.911124 Received: 2018.05.14 Accepted: 2018.07.16 Methylenetetrahydrofolate Dehydrogenase Published: 2018.11.21 1 (MTHFD1) is Underexpressed in Clear Cell Renal Cell Carcinoma Tissue and Transfection and Overexpression in Caki-1 Cells Inhibits Cell Proliferation and Increases Apoptosis Authors’ Contribution: AB Donglin He Department of Urology, Chongqing Three Gorges Central Hospital, Chongqing, Study Design A BC Zhihai Yu P.R. China Data Collection B Statistical Analysis C CD Sheng Liu Data Interpretation D BD Hong Dai Manuscript Preparation E DF Qing Xu Literature Search F Funds Collection G EF Feng Li Corresponding Author: Feng Li, e-mail: [email protected] Source of support: Departmental sources Background: The aims of this study were to investigate the expression of methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) in human tissue containing clear cell renal cell carcinoma (CCRCC) compared with normal renal tis- sue, and the effects of upregulating the expression of MTHFD1 in the human CCRCC cell line, Caki-1. Material/Methods: Tumor and adjacent normal renal tissue were obtained from 44 patients who underwent radical nephrectomy for CCRCC. Caki-1 human CCRCC cells were divided into the control group, the empty vector (EV) group, and the plasmid-treated group that overexpressed MTHFD1. MTHFD1 mRNA and protein levels were measured by quantitative real-time polymerase chain reaction (qRT-PCR) and Western blot, respectively. The cell counting kit-8 (CCK-8) assay measured cell viability. Flow cytometry evaluated apoptosis and the cell cycle. Western blot measured the protein levels of MTHFD1, Bax, Bcl-2, Akt, p53, and cyclin D1, and qRT-PCR determined the gene expression profiles. -

Mitochondrial Methylenetetrahydrofolate
Published OnlineFirst June 22, 2015; DOI: 10.1158/1541-7786.MCR-15-0117 Perspective Molecular Cancer Research Mitochondrial Methylenetetrahydrofolate Dehydrogenase (MTHFD2) Overexpression Is Associated with Tumor Cell Proliferation and Is a Novel Target for Drug Development Philip M. Tedeschi1, Alexei Vazquez2, John E. Kerrigan3, and Joseph R. Bertino4 Abstract Rapidly proliferating tumors attempt to meet the demands for sensitive to treatment with MTX. A key enzyme upregulated in nucleotide biosynthesis by upregulating folate pathways that rapidly proliferating tumors but not in normal adult cells is the provide the building blocks for pyrimidine and purine biosyn- mitochondrial enzyme methylenetetrahydrofolate dehydroge- thesis. In particular, the key role of mitochondrial folate enzymes nase (MTHFD2). This perspective outlines the rationale for spe- in providing formate for de novo purine synthesis and for provid- cific targeting of MTHFD2 and compares known and generated ing the one-carbon moiety for thymidylate synthesis has been crystal structures of MTHFD2 and closely related enzymes as a recognized in recent studies. We have shown a significant corre- molecular basis for developing therapeutic agents against lation between the upregulation of the mitochondrial folate MTHFD2. Importantly, the development of selective inhibitors enzymes, high proliferation rates, and sensitivity to the folate of mitochondrial methylenetetrahydrofolate dehydrogenase is antagonist methotrexate (MTX). Burkitt lymphoma and diffuse expected to have substantial activity, and this perspective supports large-cell lymphoma tumor specimens have the highest levels of the investigation and development of MTHFD2 inhibitors for mitochondrial folate enzyme expression and are known to be anticancer therapy. Mol Cancer Res; 13(10); 1361–6. Ó2015 AACR. Introduction this activity in embryos was shown by knocking out this gene (nmdmc) in mice.