Organic Transformations Catalyzed by Methylrhenium Trioxide Zuolin Zhu Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hydrogenation of Styrene Oxide to 2-Phenylethanol Over Nanocrystalline Ni Prepared by Ethylene Glycol Reduction Method
Hindawi Publishing Corporation International Journal of Chemical Engineering Volume 2014, Article ID 406939, 6 pages http://dx.doi.org/10.1155/2014/406939 Research Article Hydrogenation of Styrene Oxide to 2-Phenylethanol over Nanocrystalline Ni Prepared by Ethylene Glycol Reduction Method Sunil K. Kanojiya,1 G. Shukla,1 S. Sharma,1 R. Dwivedi,2 P. Sharma,2 R. Prasad,2 M. Satalkar,3 and S. N. Kane3 1 IPCA Laboratories Ltd., Indore 452003, India 2 School of Chemical Sciences, DA University, Indore 452001, India 3 Magnetic Materials Laboratory, School of Physics, DA University, Indore 452001, India Correspondence should be addressed to R. Prasad; [email protected] Received 14 February 2014; Accepted 20 May 2014; Published 17 July 2014 Academic Editor: Moshe Sheintuch Copyright © 2014 Sunil K. Kanojiya et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Nanocrystalline nickel prepared by glycol reduction method and characterized by XRD and magnetic measurements has been used as a catalyst for hydrogenation of styrene oxide to 2-phenylethanol. Effect of process variables such as particle size of the catalyst, temperature, and pressure have been optimized to achieve a maximum conversion of 98% of styrene oxide with 99% selectivity towards 2-phenylethanol. The structure of the transition state has been computed employing density functional theory −1 and using Gaussian 09 suite. The enthalpy of reaction (Δ) and activation energy ()arecalculatedtobe85.3kcal⋅mol and −1 123.03 kcal⋅mol , respectively. A tentative mechanism for the reaction is proposed according to which atomized hydrogen and styrene oxide react together over the catalyst surface to produce 2-phenylethanol. -

Enlarging Knowledge on Lager Beer Volatile Metabolites Using Multidimensional Gas Chromatography
foods Article Enlarging Knowledge on Lager Beer Volatile Metabolites Using Multidimensional Gas Chromatography Cátia Martins 1 , Tiago Brandão 2, Adelaide Almeida 3 and Sílvia M. Rocha 1,* 1 Departamento de Química & LAQV-REQUIMTE, Universidade de Aveiro, Campus Universitário Santiago, 3810-193 Aveiro, Portugal; [email protected] 2 Super Bock Group, Rua do Mosteiro, 4465-703 Leça do Balio, Portugal; [email protected] 3 Departamento de Biologia & CESAM, Universidade de Aveiro, Campus Universitário Santiago, 3810-193 Aveiro, Portugal; [email protected] * Correspondence: [email protected]; Tel.: +351-234-401-524 Received: 30 July 2020; Accepted: 6 September 2020; Published: 11 September 2020 Abstract: Foodomics, emergent field of metabolomics, has been applied to study food system processes, and it may be useful to understand sensorial food properties, among others, through foods metabolites profiling. Thus, as beer volatile components represent the major contributors for beer overall and peculiar aroma properties, this work intends to perform an in-depth profiling of lager beer volatile metabolites and to generate new data that may contribute for molecules’ identification, by using multidimensional gas chromatography. A set of lager beers were used as case-study, and 329 volatile metabolites were determined, distributed over 8 chemical families: acids, alcohols, esters, monoterpenic compounds, norisoprenoids, sesquiterpenic compounds, sulfur compounds, and volatile phenols. From these, 96 compounds are reported for the first time in the lager beer volatile composition. Around half of them were common to all beers under study. Clustering analysis allowed a beer typing according to production system: macro- and microbrewer beers. Monoterpenic and sesquiterpenic compounds were the chemical families that showed wide range of chemical structures, which may contribute for the samples’ peculiar aroma characteristics. -

BACKGROUND DOCUMENT for DYES METABOLIZED to BENZIDINE (BENZIDINE DYE CLASS)
NTP REPORT ON CARCINOGENS BACKGROUND DOCUMENT for DYES METABOLIZED TO BENZIDINE (BENZIDINE DYE CLASS) FINAL MARCH1999 Prepared for the October 30-31, 1997, Meeting ofthe Report on Carcinogens Subcommittee ofthe NTP Board ofScientific Counselors Prepared by Integrated Laboratory Systems Post Office Box 13501 Research Triangle Park, North Carolina 27709 NIEHS Contract No. N01-ES-25346 NTP Report on Carcinogens 1997 Background Document for Dyes Metabolized to Benzidine (Benzidine Dye Class) TABLE OF CONTENTS NTP Report on Carcinogens Listing for Dyes Metabolized to Benzidine (Benzidine Dye Class) ...•.......•..•.•.......•...•..•.•.•.••••.•.•.•••.••••••••••••••• 1 Listing Criteria from the Report on Carcinogens, Eighth Edition ••••.••••••.••.•.•.• 3 Supporting Information for Listing .........•...................•.•........•.•.•••.•••.•••.•••.•.••..•... 4 Table 1. Some Regulated Azo Dyes Derived From Benzidine That Have Citations in BIOSIS, CANCERLIT, EMBASE, MEDLINE, RTECS, and/or TOXLINE •.•..•..••..•.••••.•.•.•••.•..•.••••••••••••••••••.•• 5 REFERENCES •.•.•.•.•.••.•.••.•.•.......•..•..•.....•.•..•..•..•.•....•..•..•.•.••••.•.•.••.•••.••••••••••••••.••••• 8 APPENDIX A- Excerpts from IARC (1982a) and IARC Supplements (IARC, 1979; IARC, 1982b; and IARC, 1987) Benzidine .•••...•••.••••••••• A-1 APPENDIX B- Excerpts from IARC (1982a) Direct Black 38, Direct Blue 6, Direct Brown 95 ..•.••.••.••••.••.•••••••.•.••.••••••••.•.•••.•••.•••••••••••••• B-1 APPENDIX C- Excerpts from the NCI Technical Report 13-Week Subchronic Toxicity Studies of Direct -

Deoxydehydration of Vicinal Diols by Homogeneous Catalysts: a Mechanistic Overview
Deoxydehydration of vicinal diols by homogeneous royalsocietypublishing.org/journal/rsos catalysts: a mechanistic overview Review Cite this article: DeNike KA, Kilyanek SM. 2019 Kayla A. DeNike and Stefan M. Kilyanek Deoxydehydration of vicinal diols by homogeneous catalysts: a mechanistic overview. Department of Chemistry and Biochemistry, University of Arkansas, 1 University of Arkansas, Fayetteville, AR 727001, USA R. Soc. open sci. 6: 191165. http://dx.doi.org/10.1098/rsos.191165 SMK, 0000-0002-6179-2510 Received: 4 July 2019 Deoxydehydration (DODH) is an important reaction for the Accepted: 4 October 2019 upconversion of biomass-derived polyols to commodity chemicals such as alkenes and dienes. DODH can be performed Subject Category: by a variety of early metal-oxo catalysts incorporating Re, Mo and V. The varying reduction methods used in the DODH Chemistry catalytic cycle impact the product distribution, reaction Subject Areas: mechanism and the overall yield of the reaction. This review surveys the reduction methods commonly used in homogeneous inorganic chemistry/organometallic chemistry/ DODH catalyst systems and their impacts on yield and reaction green chemistry conditions. Keywords: deoxydehydration, catalysis, biomass upconversion, early metal-oxo 1. Introduction Due to the long-term implications for the climate of continuing to Author for correspondence: consume non-renewable carbon resources, such as oil, the Stefan M. Kilyanek decarbonization of the economy has become the topic of e-mail: [email protected] significant public and scientific concern [1,2]. As a result, the development of processes to produce valuable carbon-based commodity chemicals from renewable carbon feedstocks has become an important field of study [3]. -

Volatile Organic Compounds from Books
1 Measuring the emission of volatile organic compounds from books Velson Horie Research Project Manager The British Library What is happening to our books? presevation of folding endurance DP X. Zou, T. Uesaka, N. Gurnagul, Prediction of paper permanence by accelerated aging. Part I: Kinetic analysis of the 3 aging process , Cellulose, 1996, 3, 243-267. A Few Statistics •Formal beginning in 1753 as the library of The British Museum •The British Library formed in 1973 from many collections •New St Pancras building opened in 1998 •150m collection items on 640km of shelves, •£131m budget, 1900 staff 4 Additional Storage Programme - Boston Spa •7 million collection items •263 km, 12,000 tonne of stock •Reduced oxygen (16%) •Robotic book handling •What are the long term effects? 5 Preserving Newspapers •33 km of stock •5,300 tonne of stock •1.4 tonne/y VOC production •3,800 years till all evaporated 6 Major UK libraries and archives Cambridge University Library (CUL) 7m printed items The British Library (BL) 150m items National Library of Scotland (NLS) 14m items National Library of Wales (NLW) 6m printed items Oxford University Library(OULS) 11m items Trinity College Dublin Library (TCD) 4m printed items The National Archives (TNA) National Archives of Scotland (NAS) 7 Condition assessment Preservation Assessment Survey Strength Colour pH Molecular weight Furnish SurveNIR VOCs 8 The “real thing” is important to people E-books sales have been slow to take off. CafeScribe is sending every e-textbook purchaser a scratch and sniff sticker with a musty “old book” smell. By placing these stickers on their computers, they can give their e-books the same musty book smell they know and love from used textbooks. -

United States Patent (19) 11 Patent Number: 5,342,985 Herrmann Et Al
USOO5342985A United States Patent (19) 11 Patent Number: 5,342,985 Herrmann et al. (45. Date of Patent: Aug. 30, 1994 54) ORGANIC DERIVATIVES OF RHENIUM 58 Field of Search ................ 556/482, 485; 560/130, OXDES AND THER PREPARATION AND 560/219, 221, 205; 568/626, 627, 630, 655, 685, USE FOR THE METATHESS OF OLEFENS 687; 570/135, 136; 585/510,520 75 Inventors: Wolfgang A. Herrmann, Freising: Primary Examiner-Paul F. Shaver Werner Wagner, Munich; Ursula Attorney, Agent, or Firm-Connolly & Hutz Volkhardt, Freising, all of Fed. Rep. of Germany 57 ABSTRACT 73) Assignee: Hoechst AG, Frankfurt am Main, The invention relates to a process for the metathesis of Fed. Rep. of Germany olefins which comprises reacting an olefin of the for mula YCZ=CZ-(CX2),R2 (II) wherein 21 Appl. No.: 569,614 n is an integer from 1 to 28, 22 Filed: Aug. 20, 1990 X represents H or F, Y represents H or alkyl having from 1 to 10 carbon Related U.S. Application Data atoms and 63 Continuation-in-part of Ser. No. 320,404, Mar. 8, 1989, Z represents Hor a non-aromatic hydrocarbon group abandoned. having from 1 to 6 carbon atoms and R2 represents a H, alkyl, halogen, COOR3 or OR', wherein R3and 30 Foreign Application Priority Data R4 represent alkyl having from 1 to 15 carbon Dec. 10, 1988 DE Fed. Rep. of Germany ....... 384,733 atoms or phenyl which is unsubstituted or contains Jan. 31, 1989 IDE Fed. Rep. of Germany ....... 3902787 from 1 to 3 substituents or wherein R is trialkylsi Mar. -

Green Chemistry : Greener Alternatives to Synthetic Organic Transformations
Green Chemistry Greener Alternatives to Synthetic Organic Transformations V.K. Ahluwalia Alpha Science International Ltd. Oxford, U.K. Contents Preface v Parti 1. Introduction 1.1-1.10 1.1 Principles of Green Chemistry 1.1 1.2 How to Plan a Green Synthesis 1.2 Part II Green Alternatives to Synthesis Organic Transformations 2. Aqueous Phase Transformations 2.1-2.48 2.1 p-Acetylaminophenol (Tylenol) 2.1 2.2 3-Aminopyridine 2.2 2.2a Anthranilic Acid 2.3 2.3 Benzilic Acid 2.3 2.4 Benzoin 2.5 2.5 Benzotriazole 2.6 2.6 2-Benzoyl-3,5-dimethylbenzofuran 2.7 2.7 n-Butyl Bromide 2.8 2.8 Tert.Butylchloride 2.8 2.9 Chalcone (Benzalacetophenone) 2.9 2.10 Cycloheptanone 2.10 2.11 2,3-Dihydroxy Anisole (Pyrigallol Monomethyl-ether) 2.12 2.12 2,4-Dihydroxybenzoic acid (P-resorcylic Acid) 2.13 2.13 3,4-Dimethoxyphenol 2.14 2.14 2,3-Dimethyl-l-phenylpyrazol-5-one 2.15 2.15 3, 5-Dimethylpyrazole 2.16 2.16 5,5-Diphenylhydantoin 2.17 2.17 Endo-cis-1,4-endoxo-A5-cyclohexene-2,3-dicarboxylic Acid 2.18 2.18 p-Ethoxyacetanilide (Phenacetin) 2.19 2.19 6-Ethoxycarbonyl-3,5-diphenyI-2-cyclohexenone 2.20 2.20 HeteroDiels-AlderAdduct 2.22 2.21 Hippuric Acid (Benzoyl Glycine) 2.22 Viii Contents 2.22 Hydantion 2.23 2.25 3-Hydroxy-3-phenyl-2-methylene Proponamide 2.24 2.24 Iodoform 2.25 2.25 Inodole 2.26 2.26 3-(p-Methoxyphenyl)-2H-1,4-Benzoxazine 2.27 2.27 3-Methylcyclopent-2-enone 2.27 2.28 2-(2'-Methlindol-3yl)-1,4-benzoquinone 2.28 2.29 2-methyl-2-(3-oxobutyl)-1,3-cyclopentanedione 2.29 2.30 P-Naphthyl Acetate 2.30 2.31 (5-Naphthyl Methyl Ether (Nerolin) 2.31 2.32 -

Inhibition of Growth, Synthesis, and Permeability in Neurospora Crassa by Phenethyl Alcohol
JOURNAL OF BACTERIOLOGY, JUly, 1965 Vol. 90, No. I Copyright © 1965 American Society for Microbiology Printed in U.S.A. Inhibition of Growth, Synthesis, and Permeability in Neurospora crassa by Phenethyl Alcohol GABRIEL LESTER Department of Biology, Reed College, Portland, Oregon Received for publication 22 January 1965 ABSTRACT LESTER, GABRIEL (Reed College, Portland, Ore.). Inhibition of growth, synthesis, and permeability in Neurospora crassa by phenethyl alcohol. J. Bacteriol. 90: 29-37. 1965.- Inhibition of the growth of Neurospora crassa in still culture was detected at 0.05% and was complete at a level of 0.2% phenethyl alcohol (PEA). Benzyl alcohol was less in- hibitory, and 3-phenyl-1-propanol and phenol were more inhibitory, than PEA; benzyl- amine and phenethylamine were less inhibitory than the analogous hydroxylated com- pounds. Inhibition by PEA was not reversed by synthetic mixtures of purines and pyrimidines or vitamins, or by casein digests, yeast extract, or nutrient broth. The germination of conidia was inhibited by PEA, but after an exposure of 8.5 hr no loss of viability was observed. The addition of PEA to growing shake cultures caused a simul- taneous inhibition of growth and of the syntheses of ribonucleic and deoxyribonucleic acids and protein; the relationships of these compounds to mycelial dry weight and to one another were constant in growing mycelia, and PEA did not significantly affect these relationships. PEA partially inhibited the uptake of glucose, but severely re- stricted the accumulation of L-leucine, L-tryptophan, or a-aminoisobutyric acid in germinated conidia. The efflux of a-aminoisobutyric acid from germinated conidia was somewhat enhanced by PEA, but this effect was not so pronounced as the (complete) inhibition of a-aminoisobutyric acid accumulation by PEA. -

United States Patent (19) 11 4,267,375 Maasbol Et Al
United States Patent (19) 11 4,267,375 Maasbol et al. 45 May 12, 1981 54 PREPARATION OF THIOETHERS OTHER PUBLICATIONS 75) Inventors: Alfred G. Maasbol, Hamburg, Fed. I. Ruderman et al., J. Amer. Chem. Soc., 71, pp. Rep. of Germany; Lothar G. Dulog, 2264-2265, (1949). St. Martens Latem, Belgium Morrisson and Boyd, Organic Chemistry, 2nd edition, (1967), pp. 29–30. 73) Assignee: s.a. Texaco Belgium in.v., Brussels, T. Todsen et al., J. Amer. Chem. Soc., 72, pp. Belgium 4000-4002, (1950). Berichte Deutsch. Chemie, vol. 1, pp. 587-591, (1935), (21) Appl. No.: 945,273 Berlin. 22 Filed: Sep. 25, 1978 D. Gregg et al., J. Org. Chem., pp. 246-252, (1950). M. Malinovskii, Epoxides and Their Derivatives, pp. Related U.S. Application Data 131-136, (1965), Jerusalem, Daniel Davey & Co. Primary Examiner-Glennon H. Hollrah 63 Continuation of Ser. No. 703,045, Jul. 6, 1976, aban Assistant Examiner-M. C. Eakin doned. Attorney, Agent, or Firm-Carl G. Ries; Robert A. 30 Foreign Application Priority Data Kulason; Carl G. Seutter Nov. 19, 1975 GB United Kingdom ............... 47582/75 57 ABSTRACT .. 51 Int. Cl. ............................................ CO7C 149/30 Thioethers may be prepared by reacting a thiol, such as thiophenol, with an alcohol (having electron donor 52 U.S. C. ......................................... 568/57; 568/58 groups in the alpha or beta position to its hydroxyl 58 Field of Search ........................ 260/609 E, 609 R group) such as phenyl-1-hydroxy-phenethylsulfide. Re 56) References Cited action is carried out in the presence of a Lewis Acid U.S. PATENT DOCUMENTS metal halide, typically zinc chloride. -
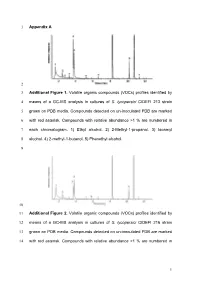
Appendix a 1 2 Additional Figure 1. Volatile Organic Compounds (Vocs
1 Appendix A 2 3 Additional Figure 1. Volatile organic compounds (VOCs) profiles identified by 4 means of a GC-MS analysis in cultures of S. lycopersici CIDEFI 213 strain 5 grown on PDB media. Compounds detected on un-inoculated PDB are marked 6 with red asterisk. Compounds with relative abundance >1 % are numbered in 7 each chromatogram. 1) Ethyl alcohol. 2) 2-Methyl-1-propanol. 3) Isoamyl 8 alcohol. 4) 2-methyl-1-butanol. 5) Phenethyl alcohol. 9 10 11 Additional Figure 2. Volatile organic compounds (VOCs) profiles identified by 12 means of a GC-MS analysis in cultures of S. lycopersici CIDEFI 216 strain 13 grown on PDB media. Compounds detected on un-inoculated PDB are marked 14 with red asterisk. Compounds with relative abundance >1 % are numbered in 1 15 each chromatogram. 1) Ethyl alcohol. 2) 2-Methyl-1-propanol. 3) Isoamyl 16 alcohol. 4) 2-methyl-1-butanol. 5) Phenethyl alcohol. 6) Furfuryl alcohol. 17 18 19 Additional Figure 3. Volatile organic compounds (VOCs) profiles identified by 20 means of a GC-MS analysis in cultures of F. fulva CIDEFI 300 strain grown on 21 PDB media. Compounds detected on un-inoculated PDB are marked with red 22 asterisk. Compounds with relative abundance >1 % are numbered in each 23 chromatogram. 6) Furfuryl alcohol. 7) Acetone. 8) Methyl trimethylacetate. 9) 24 Isoamyl alcohol. 10) 1-Octene. 11) 3-Hexanone, 4-methyl-. 12) Styrene. 13) 3- 25 Octanone. 14) Hexanoic acid, 2 ethyl-, methyl ester. 15) 2-Nonanone. 16) 26 Phenethyl alcohol. 17) No identified Nist05. 27 2 28 29 Additional Figure 4. -

(12) United States Patent (10) Patent No.: US 8,486,374 B2 Tamarkin Et Al
USOO8486374B2 (12) United States Patent (10) Patent No.: US 8,486,374 B2 Tamarkin et al. (45) Date of Patent: Jul. 16, 2013 (54) HYDROPHILIC, NON-AQUEOUS (56) References Cited PHARMACEUTICAL CARRIERS AND COMPOSITIONS AND USES U.S. PATENT DOCUMENTS 1,159,250 A 11/1915 Moulton 1,666,684 A 4, 1928 Carstens (75) Inventors: Dov Tamarkin, Maccabim (IL); Meir 1924,972 A 8, 1933 Beckert Eini, Ness Ziona (IL); Doron Friedman, 2,085,733. A T. 1937 Bird Karmei Yosef (IL); Alex Besonov, 2,390,921 A 12, 1945 Clark Rehovot (IL); David Schuz. Moshav 2,524,590 A 10, 1950 Boe Gimzu (IL); Tal Berman, Rishon 2,586.287 A 2/1952 Apperson 2,617,754 A 1 1/1952 Neely LeZiyyon (IL); Jorge Danziger, Rishom 2,767,712 A 10, 1956 Waterman LeZion (IL); Rita Keynan, Rehovot (IL); 2.968,628 A 1/1961 Reed Ella Zlatkis, Rehovot (IL) 3,004,894 A 10/1961 Johnson et al. 3,062,715 A 11/1962 Reese et al. 3,067,784. A 12/1962 Gorman (73) Assignee: Foamix Ltd., Rehovot (IL) 3,092.255. A 6, 1963 Hohman 3,092,555 A 6, 1963 Horn 3,141,821 A 7, 1964 Compeau (*) Notice: Subject to any disclaimer, the term of this 3,142,420 A 7/1964 Gawthrop patent is extended or adjusted under 35 3,144,386 A 8/1964 Brightenback U.S.C. 154(b) by 1180 days. 3,149,543 A 9, 1964 Naab 3,154,075 A 10, 1964 Weckesser 3,178,352 A 4, 1965 Erickson (21) Appl. -

(12) Patent Application Publication (10) Pub. No.: US 2011/0028732 A1 Trauth Et Al
US 2011 0028732A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2011/0028732 A1 Trauth et al. (43) Pub. Date: Feb. 3, 2011 (54) NITRATED HYDROCARBONS, (86). PCT No.: PCT/US2009/0399.01 DERIVATIVES, AND PROCESSES FOR THEIR MANUFACTURE S371 (c)(1), (2), (4) Date: Sep. 27, 2010 (75) Inventors: Daniel M. Trauth, Crystal Lake, IL Related U.S. Application Data (US); George D. Green, Cary, IL (US); Raymond J. Swedo, Mt. (60) Eyal application No. 61/045.380, filed on Apr. Prospect, IL (US); Richard L. s James, Eros, LA (US); Ian A. Publication Classification Tomlinson, Midland, MI (US) (51) Int. Cl. C07D 263/04 (2006.01) Correspondence Address: C07C 205/05 (2006.01) The Dow Chemical Company C07C 205/01 (2006.01) P.O. BOX 1967, 2040 Dow Center CD7C 205/06 (2006.01) Midland, MI 48641 (US) C07C 215/02 (2006.01) C07C 239/08 (2006.01) (73) Assignees: ANGUS CHEMICAL (52) U.S. Cl. ......... 548/215; 7.3,So COMPANY, Buffalo Grove, IL s s (US): GLOBAL (57) ABSTRACT TECHNOLOGIES INC. , MidlandM1Clland, Provided is a process for the formation of nitrated compounds MI (US) by the nitration of hydrocarbon compounds with- dilute- - nitric acid. Also provided are processes for preparing industrially (21) Appl. No.: 12/934,817 useful downstream derivatives of the nitrated compounds, as well as novel nitrated compounds and derivatives, and meth (22) PCT Filed: Apr. 8, 2009 ods of using the derivatives in various applications. US 2011/0028732 A1 Feb. 3, 2011 NITRATED HYDROCARBONS, 0009. The invention further provides methods of using the DERIVATIVES, AND PROCESSES FOR THEIR nitrated hydrocarbons and derivatives thereof in various MANUFACTURE applications.