Statistical Mechanics I: Final Exam Review Solution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Competition Between Paramagnetism and Diamagnetism in Charged
Competition between paramagnetism and diamagnetism in charged Fermi gases Xiaoling Jian, Jihong Qin, and Qiang Gu∗ Department of Physics, University of Science and Technology Beijing, Beijing 100083, China (Dated: November 10, 2018) The charged Fermi gas with a small Lande-factor g is expected to be diamagnetic, while that with a larger g could be paramagnetic. We calculate the critical value of the g-factor which separates the dia- and para-magnetic regions. In the weak-field limit, gc has the same value both at high and low temperatures, gc = 1/√12. Nevertheless, gc increases with the temperature reducing in finite magnetic fields. We also compare the gc value of Fermi gases with those of Boltzmann and Bose gases, supposing the particle has three Zeeman levels σ = 1, 0, and find that gc of Bose and Fermi gases is larger and smaller than that of Boltzmann gases,± respectively. PACS numbers: 05.30.Fk, 51.60.+a, 75.10.Lp, 75.20.-g I. INTRODUCTION mK and 10 µK, respectively. The ions can be regarded as charged Bose or Fermi gases. Once the quantum gas Magnetism of electron gases has been considerably has both the spin and charge degrees of freedom, there studied in condensed matter physics. In magnetic field, arises the competition between paramagnetism and dia- the magnetization of a free electron gas consists of two magnetism, as in electrons. Different from electrons, the independent parts. The spin magnetic moment of elec- g-factor for different magnetic ions is diverse, ranging trons results in the paramagnetic part (the Pauli para- from 0 to 2. -

Solid State Physics 2 Lecture 5: Electron Liquid
Physics 7450: Solid State Physics 2 Lecture 5: Electron liquid Leo Radzihovsky (Dated: 10 March, 2015) Abstract In these lectures, we will study itinerate electron liquid, namely metals. We will begin by re- viewing properties of noninteracting electron gas, developing its Greens functions, analyzing its thermodynamics, Pauli paramagnetism and Landau diamagnetism. We will recall how its thermo- dynamics is qualitatively distinct from that of a Boltzmann and Bose gases. As emphasized by Sommerfeld (1928), these qualitative di↵erence are due to the Pauli principle of electons’ fermionic statistics. We will then include e↵ects of Coulomb interaction, treating it in Hartree and Hartree- Fock approximation, computing the ground state energy and screening. We will then study itinerate Stoner ferromagnetism as well as various response functions, such as compressibility and conduc- tivity, and screening (Thomas-Fermi, Debye). We will then discuss Landau Fermi-liquid theory, which will allow us understand why despite strong electron-electron interactions, nevertheless much of the phenomenology of a Fermi gas extends to a Fermi liquid. We will conclude with discussion of electrons on the lattice, treated within the Hubbard and t-J models and will study transition to a Mott insulator and magnetism 1 I. INTRODUCTION A. Outline electron gas ground state and excitations • thermodynamics • Pauli paramagnetism • Landau diamagnetism • Hartree-Fock theory of interactions: ground state energy • Stoner ferromagnetic instability • response functions • Landau Fermi-liquid theory • electrons on the lattice: Hubbard and t-J models • Mott insulators and magnetism • B. Background In these lectures, we will study itinerate electron liquid, namely metals. In principle a fully quantum mechanical, strongly Coulomb-interacting description is required. -

Particle Characterisation In
LIFE SCIENCE I TECHNICAL BULLETIN ISSUE N°11 /JULY 2008 PARTICLE CHARACTERISATION IN EXCIPIENTS, DRUG PRODUCTS AND DRUG SUBSTANCES AUTHOR: HILDEGARD BRÜMMER, PhD, CUSTOMER SERVICE MANAGER, SGS LIFE SCIENCE SERVICES, GERMANY Particle characterization has significance in many industries. For the pharmaceutical industry, particle size impacts products in two ways: as an influence on drug performance and as an indicator of contamination. This article will briefly examine how particle size impacts both, and review the arsenal of methods for measuring and tracking particle size. Furthermore, examples of compromised product quality observed within our laboratories illustrate why characterization is so important. INDICATOR OF CONTAMINATION Controlling the limits of contaminating • Adverse indirect reactions: particles Particle characterisation of drug particles is critical for injectable are identified by the immune system as substances, drug products and excipients (parenteral) solutions. Particle foreign material and immune reaction is an important factor in R&D, production contamination of solutions can potentially might impose secondary effects. and quality control of pharmaceuticals. have the following results: It is becoming increasingly important for In order to protect a patient and to compliance with requirements of FDA and • Adverse direct reactions: e.g. particles guarantee a high quality product, several European Health Authorities. are distributed via the blood in the body chapters in the compendia (USP, EP, JP) and cause toxicity to specific tissues or describe techniques for characterisation organs, or particles of a given size can of limits. Some of the most relevant cause a physiological effect blocking blood chapters are listed in Table 1. flow e.g. in the lungs. -

Chapter 3 Bose-Einstein Condensation of an Ideal
Chapter 3 Bose-Einstein Condensation of An Ideal Gas An ideal gas consisting of non-interacting Bose particles is a ¯ctitious system since every realistic Bose gas shows some level of particle-particle interaction. Nevertheless, such a mathematical model provides the simplest example for the realization of Bose-Einstein condensation. This simple model, ¯rst studied by A. Einstein [1], correctly describes important basic properties of actual non-ideal (interacting) Bose gas. In particular, such basic concepts as BEC critical temperature Tc (or critical particle density nc), condensate fraction N0=N and the dimensionality issue will be obtained. 3.1 The ideal Bose gas in the canonical and grand canonical ensemble Suppose an ideal gas of non-interacting particles with ¯xed particle number N is trapped in a box with a volume V and at equilibrium temperature T . We assume a particle system somehow establishes an equilibrium temperature in spite of the absence of interaction. Such a system can be characterized by the thermodynamic partition function of canonical ensemble X Z = e¡¯ER ; (3.1) R where R stands for a macroscopic state of the gas and is uniquely speci¯ed by the occupa- tion number ni of each single particle state i: fn0; n1; ¢ ¢ ¢ ¢ ¢ ¢g. ¯ = 1=kBT is a temperature parameter. Then, the total energy of a macroscopic state R is given by only the kinetic energy: X ER = "ini; (3.2) i where "i is the eigen-energy of the single particle state i and the occupation number ni satis¯es the normalization condition X N = ni: (3.3) i 1 The probability -

Grand-Canonical Ensemble
PHYS4006: Thermal and Statistical Physics Lecture Notes (Unit - IV) Open System: Grand-canonical Ensemble Dr. Neelabh Srivastava (Assistant Professor) Department of Physics Programme: M.Sc. Physics Mahatma Gandhi Central University Semester: 2nd Motihari-845401, Bihar E-mail: [email protected] • In microcanonical ensemble, each system contains same fixed energy as well as same number of particles. Hence, the system dealt within this ensemble is a closed isolated system. • With microcanonical ensemble, we can not deal with the systems that are kept in contact with a heat reservoir at a given temperature. 2 • In canonical ensemble, the condition of constant energy is relaxed and the system is allowed to exchange energy but not the particles with the system, i.e. those systems which are not isolated but are in contact with a heat reservoir. • This model could not be applied to those processes in which number of particle varies, i.e. chemical process, nuclear reactions (where particles are created and destroyed) and quantum process. 3 • So, for the method of ensemble to be applicable to such processes where number of particles as well as energy of the system changes, it is necessary to relax the condition of fixed number of particles. 4 • Such an ensemble where both the energy as well as number of particles can be exchanged with the heat reservoir is called Grand Canonical Ensemble. • In canonical ensemble, T, V and N are independent variables. Whereas, in grand canonical ensemble, the system is described by its temperature (T),volume (V) and chemical potential (μ). 5 • Since, the system is not isolated, its microstates are not equally probable. -

The Grand Canonical Ensemble
University of Central Arkansas The Grand Canonical Ensemble Stephen R. Addison Directory ² Table of Contents ² Begin Article Copyright °c 2001 [email protected] Last Revision Date: April 10, 2001 Version 0.1 Table of Contents 1. Systems with Variable Particle Numbers 2. Review of the Ensembles 2.1. Microcanonical Ensemble 2.2. Canonical Ensemble 2.3. Grand Canonical Ensemble 3. Average Values on the Grand Canonical Ensemble 3.1. Average Number of Particles in a System 4. The Grand Canonical Ensemble and Thermodynamics 5. Legendre Transforms 5.1. Legendre Transforms for two variables 5.2. Helmholtz Free Energy as a Legendre Transform 6. Legendre Transforms and the Grand Canonical Ensem- ble 7. Solving Problems on the Grand Canonical Ensemble Section 1: Systems with Variable Particle Numbers 3 1. Systems with Variable Particle Numbers We have developed an expression for the partition function of an ideal gas. Toc JJ II J I Back J Doc Doc I Section 2: Review of the Ensembles 4 2. Review of the Ensembles 2.1. Microcanonical Ensemble The system is isolated. This is the ¯rst bridge or route between mechanics and thermodynamics, it is called the adiabatic bridge. E; V; N are ¯xed S = k ln (E; V; N) Toc JJ II J I Back J Doc Doc I Section 2: Review of the Ensembles 5 2.2. Canonical Ensemble System in contact with a heat bath. This is the second bridge between mechanics and thermodynamics, it is called the isothermal bridge. This bridge is more elegant and more easily crossed. T; V; N ¯xed, E fluctuates. -

2. Classical Gases
2. Classical Gases Our goal in this section is to use the techniques of statistical mechanics to describe the dynamics of the simplest system: a gas. This means a bunch of particles, flying around in a box. Although much of the last section was formulated in the language of quantum mechanics, here we will revert back to classical mechanics. Nonetheless, a recurrent theme will be that the quantum world is never far behind: we’ll see several puzzles, both theoretical and experimental, which can only truly be resolved by turning on ~. 2.1 The Classical Partition Function For most of this section we will work in the canonical ensemble. We start by reformu- lating the idea of a partition function in classical mechanics. We’ll consider a simple system – a single particle of mass m moving in three dimensions in a potential V (~q ). The classical Hamiltonian of the system3 is the sum of kinetic and potential energy, p~ 2 H = + V (~q ) 2m We earlier defined the partition function (1.21) to be the sum over all quantum states of the system. Here we want to do something similar. In classical mechanics, the state of a system is determined by a point in phase space.Wemustspecifyboththeposition and momentum of each of the particles — only then do we have enough information to figure out what the system will do for all times in the future. This motivates the definition of the partition function for a single classical particle as the integration over phase space, 1 3 3 βH(p,q) Z = d qd pe− (2.1) 1 h3 Z The only slightly odd thing is the factor of 1/h3 that sits out front. -

Particle Number Concentrations and Size Distribution in a Polluted Megacity
https://doi.org/10.5194/acp-2020-6 Preprint. Discussion started: 17 February 2020 c Author(s) 2020. CC BY 4.0 License. Particle number concentrations and size distribution in a polluted megacity: The Delhi Aerosol Supersite study Shahzad Gani1, Sahil Bhandari2, Kanan Patel2, Sarah Seraj1, Prashant Soni3, Zainab Arub3, Gazala Habib3, Lea Hildebrandt Ruiz2, and Joshua S. Apte1 1Department of Civil, Architectural and Environmental Engineering, The University of Texas at Austin, Texas, USA 2McKetta Department of Chemical Engineering, The University of Texas at Austin, Texas, USA 3Department of Civil Engineering, Indian Institute of Technology Delhi, New Delhi, India Correspondence: Joshua S. Apte ([email protected]), Lea Hildebrandt Ruiz ([email protected]) Abstract. The Indian national capital, Delhi, routinely experiences some of the world's highest urban particulate matter concentrations. While fine particulate matter (PM2.5) mass concentrations in Delhi are at least an order of magnitude higher than in many western cities, the particle number (PN) concentrations are not similarly elevated. Here we report on 1.25 years of highly 5 time resolved particle size distributions (PSD) data in the size range of 12–560 nm. We observed that the large number of accumulation mode particles—that constitute most of the PM2.5 mass—also contributed substantially to the PN concentrations. The ultrafine particles (UFP, Dp <100 nm) fraction of PN was higher during the traffic rush hours and for daytimes of warmer seasons—consistent with traffic and nucleation events being major sources of urban UFP. UFP concentrations were found to be relatively lower during periods with some of the highest mass concentrations. -
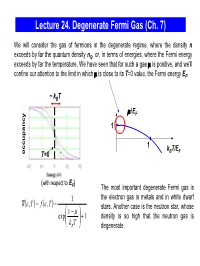
Lecture 24. Degenerate Fermi Gas (Ch
Lecture 24. Degenerate Fermi Gas (Ch. 7) We will consider the gas of fermions in the degenerate regime, where the density n exceeds by far the quantum density nQ, or, in terms of energies, where the Fermi energy exceeds by far the temperature. We have seen that for such a gas μ is positive, and we’ll confine our attention to the limit in which μ is close to its T=0 value, the Fermi energy EF. ~ kBT μ/EF 1 1 kBT/EF occupancy T=0 (with respect to E ) F The most important degenerate Fermi gas is 1 the electron gas in metals and in white dwarf nε()(),, T= f ε T = stars. Another case is the neutron star, whose ε⎛ − μ⎞ exp⎜ ⎟ +1 density is so high that the neutron gas is ⎝kB T⎠ degenerate. Degenerate Fermi Gas in Metals empty states ε We consider the mobile electrons in the conduction EF conduction band which can participate in the charge transport. The band energy is measured from the bottom of the conduction 0 band. When the metal atoms are brought together, valence their outer electrons break away and can move freely band through the solid. In good metals with the concentration ~ 1 electron/ion, the density of electrons in the electron states electron states conduction band n ~ 1 electron per (0.2 nm)3 ~ 1029 in an isolated in metal electrons/m3 . atom The electrons are prevented from escaping from the metal by the net Coulomb attraction to the positive ions; the energy required for an electron to escape (the work function) is typically a few eV. -

Chapter 13 Ideal Fermi
Chapter 13 Ideal Fermi gas The properties of an ideal Fermi gas are strongly determined by the Pauli principle. We shall consider the limit: k T µ,βµ 1, B � � which defines the degenerate Fermi gas. In this limit, the quantum mechanical nature of the system becomes especially important, and the system has little to do with the classical ideal gas. Since this chapter is devoted to fermions, we shall omit in the following the subscript ( ) that we used for the fermionic statistical quantities in the previous chapter. − 13.1 Equation of state Consider a gas ofN non-interacting fermions, e.g., electrons, whose one-particle wave- functionsϕ r(�r) are plane-waves. In this case, a complete set of quantum numbersr is given, for instance, by the three cartesian components of the wave vector �k and thez spin projectionm s of an electron: r (k , k , k , m ). ≡ x y z s Spin-independent Hamiltonians. We will consider only spin independent Hamiltonian operator of the type ˆ 3 H= �k ck† ck + d r V(r)c r†cr , �k � where thefirst and the second terms are respectively the kinetic and th potential energy. The summation over the statesr (whenever it has to be performed) can then be reduced to the summation over states with different wavevectork(p=¯hk): ... (2s + 1) ..., ⇒ r � �k where the summation over the spin quantum numberm s = s, s+1, . , s has been taken into account by the prefactor (2s + 1). − − 159 160 CHAPTER 13. IDEAL FERMI GAS Wavefunctions in a box. We as- sume that the electrons are in a vol- ume defined by a cube with sidesL x, Ly,L z and volumeV=L xLyLz. -

Statistical Physics 06/07
STATISTICAL PHYSICS 06/07 Quantum Statistical Mechanics Tutorial Sheet 3 The questions that follow on this and succeeding sheets are an integral part of this course. The code beside each question has the following significance: • K: key question – explores core material • R: review question – an invitation to consolidate • C: challenge question – going beyond the basic framework of the course • S: standard question – general fitness training! 3.1 Particle Number Fluctuations for Fermions [s] (a) For a single fermion state in the grand canonical ensemble, show that 2 h(∆nj) i =n ¯j(1 − n¯j) wheren ¯j is the mean occupancy. Hint: You only need to use the exclusion principle not the explict form ofn ¯j. 2 How is the fact that h(∆nj) i is not in general small compared ton ¯j to be reconciled with the sharp values of macroscopic observables? (b) For a gas of noninteracting particles in the grand canonical ensemble, show that 2 X 2 h(∆N) i = h(∆nj) i j (you will need to invoke that nj and nk are uncorrelated in the GCE for j 6= k). Hence show that for noninteracting Fermions Z h(∆N)2i = g() f(1 − f) d follows from (a) where f(, µ) is the F-D distribution and g() is the density of states. (c) Show that for low temperatures f(1 − f) is sharply peaked at = µ, and hence that 2 h∆N i ' kBT g(F ) where F = µ(T = 0) R ∞ ex dx [You may use without proof the result that −∞ (ex+1)2 = 1.] 3.2 Entropy of the Ideal Fermi Gas [C] The Grand Potential for an ideal Fermi is given by X Φ = −kT ln [1 + exp β(µ − j)] j Show that for Fermions X Φ = kT ln(1 − pj) , j where pj = f(j) is the probability of occupation of the state j. -

Appendix a Review of Probability Distribution
Appendix A Review of Probability Distribution In this appendix we review some fundamental concepts of probability theory. After deriving in Sect. A.1 the binomial probability distribution, in Sect. A.2 we show that for systems described by a large number of random variables this distribution tends to a Gaussian function. Finally, in Sect. A.3 we define moments and cumulants of a generic probability distribution, showing how the central limit theorem can be easily derived. A.1 Binomial Distribution Consider an ideal gas at equilibrium, composed of N particles, contained in a box of volume V . Isolating within V a subsystem of volume v, as the gas is macroscop- ically homogeneous, the probability to find a particle within v equals p = v/V , while the probability to find it in a volume V − v is q = 1 − p. Accordingly, the probability to find any one given configuration (i.e. assuming that particles could all be distinguished from each other), with n molecules in v and the remaining (N − n) within (V − v), would be equal to pnqN−n, i.e. the product of the respective prob- abilities.1 However, as the particles are identical to each other, we have to multiply this probability by the Tartaglia pre-factor, that us the number of possibilities of 1Here, we assume that finding a particle within v is not correlated with finding another, which is true in ideal gases, that are composed of point-like molecules. In real fluids, however, since molecules do occupy an effective volume, we should consider an excluded volume effect.