Population Sequencing Enhances Understanding of Tea Plant Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Camel/A Sinensis L. (Cultivated Tea) and Its Wild Relatives Based on Randomly Amplified Polymorphic DNA and Organelle-Specific STS
Heredity 78(1997)603—611 Received 11 July 1996 An assessment of genetic diversity among Camel/a sinensis L. (cultivated tea) and its wild relatives based on randomly amplified polymorphic DNA and organelle-specific STS FRANCIS N. WACHIRA, WAYNE POWELL & ROBBIE WAUGH* Department of Cell and Mo/ecu/ar Genetics, Scottish Crop Research Institute, lnvergowrie, Dundee DD2 5DA, Scotland, U.K. Membersof the genus Camellia interbreed relatively freely and several natural species hybrids exist. Species introgression into the cultivated germplasm of tea, Camellia sinensis L. (0. Kuntz), from related Camellia species has been postulated, and it is thought that teas currently under cultivation are not archetypal varieties. Randomly amplified polymorphic DNAs (RAPDs) and organelle-specific polymerase chain reactions were used to establish the affin- ities among cultivated tea and its wild relatives. The measures of similarity obtained indicated that RAPDs were taxonomically informative in Came/ha, and the species relationships revealed were generally consistent with those obtained using morphological, compatibility and terpenoid affinities. Species-specific RAPD products and products potentially diagnostic of introgressive hybridization into the cultivated gene pool were identified. The organellar genomes were remarkably conserved, with polymorphism detected in only one of four noncoding regions in the chioroplast and mitochondrial genomes. Keywords: Camelliaspp., gene introgression,phenetics, randomly amplified polymorphic DNA, similarity. Introduction several, including C. taliensis, C. grandibractiata, C. kwangsiensis, C. gymnogyna, C. crassicolumna, C. Afundamental goal of germplasm collection and tachangensis, C. ptilophylla and C. irrawadiensis, are conservation is the understanding of genetic rela- already used in parts of Asia (Chang & Bartholo- tionships within and between the species of concern. -

P020110307527551165137.Pdf
CONTENT 1.MESSAGE FROM DIRECTOR …………………………………………………………………………………………………………………………………………………… 03 2.ORGANIZATION STRUCTURE …………………………………………………………………………………………………………………………………………………… 05 3.HIGHLIGHTS OF ACHIEVEMENTS …………………………………………………………………………………………………………………………………………… 06 Coexistence of Conserve and Research----“The Germplasm Bank of Wild Species ” services biodiversity protection and socio-economic development ………………………………………………………………………………………………………………………………………………… 06 The Structure, Activity and New Drug Pre-Clinical Research of Monoterpene Indole Alkaloids ………………………………………… 09 Anti-Cancer Constituents in the Herb Medicine-Shengma (Cimicifuga L) ……………………………………………………………………………… 10 Floristic Study on the Seed Plants of Yaoshan Mountain in Northeast Yunnan …………………………………………………………………… 11 Higher Fungi Resources and Chemical Composition in Alpine and Sub-alpine Regions in Southwest China ……………………… 12 Research Progress on Natural Tobacco Mosaic Virus (TMV) Inhibitors…………………………………………………………………………………… 13 Predicting Global Change through Reconstruction Research of Paleoclimate………………………………………………………………………… 14 Chemical Composition of a traditional Chinese medicine-Swertia mileensis……………………………………………………………………………… 15 Mountain Ecosystem Research has Made New Progress ………………………………………………………………………………………………………… 16 Plant Cyclic Peptide has Made Important Progress ………………………………………………………………………………………………………………… 17 Progresses in Computational Chemistry Research ………………………………………………………………………………………………………………… 18 New Progress in the Total Synthesis of Natural Products ……………………………………………………………………………………………………… -

'Camellia T'. Synonym for 'Donckelaeri'. (Masayoshi). TC Cole
T. T. Fendig. 1951, American Camellia Yearbook, p.77 as ‘Camellia T’. Synonym for ‘Donckelaeri’. (Masayoshi). T.C. Cole. Trewidden Estate Nursery, 1995, Retail Camellia List, p.8. Abbreviation for Thomas Cornelius Cole. T.C. Patin. (C.japonica) SCCS., 1976, Camellia Nomenclature, p.147: Light red. Very large, full, semi- double with irregular, large petals and a spray of large stamens. Originated in USA by T.C. Patin, Hammond, Louisiana. Sport: T.C. Patin Variegated. T.C. Patin Variegated. (C.japonica), SCCS., 1976, Camellia Nomenclature, p.147 as ‘T.C. Patin Var.’: A virus variegated form of T.C. Patin - Light red blotched white. Originated in USA by T.C. Patin, Hammond, Louisiana. T.D. Wipper. Nagoya Camellia Society Bulletin, 1992, No.25. Synonym for Dave’s Weeper. T.G. Donkelari. Lindo Nurseries Price List, 1949, p.7. Synonym for ‘Donckelaeri’. (Masayoshi). T.K. Blush. (C.japonica) Wilmot, 1943, Camellia Variety Classification Report, 1943, p.14: A light pink sport of T.K. Variegated. Originated in USA. Synonym: ‘T.K. Pink’. T.K. Number 4. Florida Nursery and Landscaping Co. Catalogue, 1948 as ‘T.K. No.4’. Synonym for T.K. Variegated. T.K. Pink. Morris, 1954, RHS., The Rhododendron and Camellia Yearbook, p.113. Synonym for T.K. Blush. T.K. Red. Semmes Nursery Catalogue, 1942-1943, p.21. Synonym for T.K. Variegated Red. T.K. Variegata. Kiyono Nursery Catalogue, 1942-1943. Synonym for T.K. Variegated. T.K. Variegated. (C.japonica) Kiyono Overlook Nursery Catalogue, 1934, p.14: Semi-double. Light pink edged dark pink. Gerbing’s Azalea Gardens Catalogue, 1938-1939: Semi-double, white flowers striped pink, rose and lavender, some flowers solid colour, purple and pink. -

Patterns of Flammability Across the Vascular Plant Phylogeny, with Special Emphasis on the Genus Dracophyllum
Lincoln University Digital Thesis Copyright Statement The digital copy of this thesis is protected by the Copyright Act 1994 (New Zealand). This thesis may be consulted by you, provided you comply with the provisions of the Act and the following conditions of use: you will use the copy only for the purposes of research or private study you will recognise the author's right to be identified as the author of the thesis and due acknowledgement will be made to the author where appropriate you will obtain the author's permission before publishing any material from the thesis. Patterns of flammability across the vascular plant phylogeny, with special emphasis on the genus Dracophyllum A thesis submitted in partial fulfilment of the requirements for the Degree of Doctor of philosophy at Lincoln University by Xinglei Cui Lincoln University 2020 Abstract of a thesis submitted in partial fulfilment of the requirements for the Degree of Doctor of philosophy. Abstract Patterns of flammability across the vascular plant phylogeny, with special emphasis on the genus Dracophyllum by Xinglei Cui Fire has been part of the environment for the entire history of terrestrial plants and is a common disturbance agent in many ecosystems across the world. Fire has a significant role in influencing the structure, pattern and function of many ecosystems. Plant flammability, which is the ability of a plant to burn and sustain a flame, is an important driver of fire in terrestrial ecosystems and thus has a fundamental role in ecosystem dynamics and species evolution. However, the factors that have influenced the evolution of flammability remain unclear. -

NLI Recommended Plant List for the Mountains
NLI Recommended Plant List for the Mountains Notable Features Requirement Exposure Native Hardiness USDA Max. Mature Height Max. Mature Width Very Wet Very Dry Drained Moist &Well Occasionally Dry Botanical Name Common Name Recommended Cultivars Zones Tree Deciduous Large (Height: 40'+) Acer rubrum red maple 'October Glory'/ 'Red Sunset' fall color Shade/sun x 2-9 75' 45' x x x fast growing, mulit-stemmed, papery peeling Betula nigra river birch 'Heritage® 'Cully'/ 'Dura Heat'/ 'Summer Cascade' bark, play props Shade/part sun x 4-8 70' 60' x x x Celtis occidentalis hackberry tough, drought tolerant, graceful form Full sun x 2-9 60' 60' x x x Fagus grandifolia american beech smooth textured bark, play props Shade/part sun x 3-8 75' 60' x x Fraxinus americana white ash fall color Full sun/part shade x 3-9 80' 60' x x x Ginkgo biloba ginkgo; maidenhair tree 'Autumn Gold'/ 'The President' yellow fall color Full sun 3-9 70' 40' x x good dappled shade, fall color, quick growing, Gleditsia triacanthos var. inermis thornless honey locust Shademaster®/ Skyline® salt tolerant, tolerant of acid, alkaline, wind. Full sun/part shade x 3-8 75' 50' x x Liriodendron tulipifera tulip poplar fall color, quick growth rate, play props, Full sun x 4-9 90' 50' x Platanus x acerifolia sycamore, planetree 'Bloodgood' play props, peeling bark Full sun x 4-9 90' 70' x x x Quercus palustris pin oak play props, good fall color, wet tolerant Full sun x 4-8 80' 50' x x x Tilia cordata Little leaf Linden, Basswood 'Greenspire' Full sun/part shade 3-7 60' 40' x x Ulmus -

Camellia Sinensis (L.) Kuntze (7)
“As Primeiras Camélias Asiáticas a Chegarem a Portugal e à Europa”. Armando Oliveira António Sanches (1623), Planisfério. 1 O género Camellia L. está praticamente confinado ao sul da China (80% de todas as espécies) e à região do sul da Ásia que inclui as Filipinas e as zonas do noroeste do arquipélago da Indonésia, com a inclusão do Japão e partes da Coreia. Estima-se que praticamente 20% das espécies de Camellia se encontram no Vietname. A região fitogeográfica do sul da Ásia é composta pela China, Laos, Mianmar (ex-Birmânia), Tailândia, Camboja e Vietname. 1 (Huang et al., 2016) 106 • A proposta taxonómica de Linnaeus (1835), “Sistema Natura”, permitiu-nos obter uma mais fácil e rápida identificação das espécies. • Baseia-se numa classificação dita binomial que atribui nomes compostos por duas palavras, quase sempre recorrendo ao latim. Adaptado de Fairy Lake Botanical Garden Flora (2018) 2 Reino Filo Classe Ordem Família Género Espécies/Variedades Cultivares Camellia caudata Wall. (11) Camellia drupifera Lour. (4) Dicotiledóneas Antófitas Camellia euryoides Lindl. (7) Vegetal (a semente Ericales (25) Theaceaes (12) Camellia (102+40) (que dão flor) contém 2 ou mais Camellia japonica L. cotilédones) Camellia kissi Wall. (11) Camellia oleifera Abel (6) Camellia rosaeflora Hook. (1) Camellia sasanqua Thunb. Camellia sinensis (L.) Kuntze (7) • A 1ª parte do nome é referente ao género da espécie em causa e a 2ª parte identifica a espécie dentro de um determinado género. Adaptado de Fairy Lake Botanical Garden Flora (2018) 2 Ordem Família -

Ornamental Shrubs and Groundcovers
ORNAMENTAL SHRUBS AND GROUNDCOVERS University of California Cooperative Extension Farm and Home Advisors 1031 S. Mt. Vernon Avenue ● Bakersfield, CA 93307 This list is intended to be a guide. Plant availability may change and improved varieties may come to market. Quantitative data do not exist for plant water use for most species. Sensitivity to possible plant allergens varies among people. AUTHOR John Karlik, Advisor, Environmental Horticulture/Environmental Science and the following: Peter Brown, Earth Landscape, Ridgecrest, CA John Karnes, Klassen Corporation Robert Martin, North of the River Recreation and Park District Mitchell Perez, Kern High School District Steph Sanders, North of the River Recreation and Park District Alfonso Valadez, Kern High School District Revised and expanded, August 2020 REFERENCES Duffield, Mary R. and Jones, Warren D. 1981. Plants for Dry Climates: How to Select, Grow and Enjoy. H.P. Publishing, Tucson, AZ Perry, Bob. 1981. Trees and Shrubs for Dry California Landscapes. Land Design Publishing, San Dimas, CA Sunset New Western Garden Book The University of California, Division of Agriculture and Natural Resources (UC ANR) prohibits discrimination against or harassment of any person employed by or seeking employment with UC ANR on the basis of race, color, national origin, religion, sex, gender, gender expression, gender identity, pregnancy (which includes pregnancy, childbirth, and medical conditions related to pregnancy or childbirth), physical or mental disability, medical condition (cancer-related or genetic characteristics), genetic information (including family medical history), ancestry, marital status, age, sexual orientation, citizenship, status as a protected veteran or service in the uniformed services (as defined by the Uniformed Services Employment and Reemployment Rights Act of 1994 [USERRA]), as well as state military and naval service. -

Camellia Queen of the Southern Garden
PLANT GUIDES CAMELLIA Queen of the Southern Garden hen Harry P. Leu gave his gardens to and newly discovered species. Thousands of camellias the City of Orlando in 1961, it was said are what make Leu Gardens unique and the array of that his Camellia assortment contained shapes, colors and sizes provide outstanding color from more than 1,500 individual plants. December through March. Guests are encouraged to WToday the internationally recognized camellia collection visit during this time to enjoy the beautiful Central continues to expand and include many historic varieties Florida weather and the Leu’s garden. Camellia sasanqua: ‘Alabama Beauty’ ‘Cotton Candy’ ‘Daydream’ ‘Fuji-no-mine’ ‘Mine-no-yuki’ Deep Pink Pink Pink and White White White ‘Pink Dauphin’ ‘Setsugekka’ ‘Cleopatra’ ‘Maiden’s Blush’ ‘Stephanie Golden’ Pink White Pink Blush Pink Hot Pink Camellias — a Primer here are tens of thousands of registered Camellias at Leu Gardens cultivars of camellias and there are Camellia sasanqua Camellia japonica more than 200 different species. n Small, often simple flowers n Very large flowers TThey are all originally native to eastern and southeastern Asia with most native to China. n Wider, open type of growth n Upright growth habit The majority of Leu Gardens’ collection n Full sun to partial shade n Prefers partial to full shade consists of cultivars of Camellia japonica and n Bloom October through n Bloom late December Camellia sasanqua. Sasanqua camellias bloom December through March earlier than the C. japonica types (often from n Nearly all fragrant n Most varieties not fragrant October through December) and most prefer full sun. -

International Camellia Journal 2017
International Camellia Journal 2017 An official publication of the International Camellia Society Journal Number 49 ISSN 0159-656X International Camellia Journal 2017 No. 49 International Camellia Society Congress 2018 Nantes, France, March 25 to 29 Pre-Congress Tour March 21-25 Gardens in Brittany Aims of the International Camellia Society Congress Registration March 25 in Nantes To foster the love of camellias throughout the world and maintain and increase their popularity Post-Congress Tours To undertake historical, scientific and horticultural research in connection with camellias Tour A March 29 to 31 Normandy, including World War II To co-operate with all national and regional camellia societies and with other horticultural societies sites To disseminate information concerning camellias by means of bulletins and other publications To encourage a friendly exchange between camellia enthusiasts of all nationalities Tour B March 29 to 31 Gardens and nurseries in southwest France Major dates in the International Camellia Society calendar Reassemble in Paris April 1 to 2, including visit to Versailles International Camellia Society Congresses 2018 - Nantes, Brittany, France. 2020 - Goto City, Japan. 2022 - Italy ISSN 0159-656X Published in 2017 by the International Camellia Society. © The International Camellia Society unless otherwise stated 3 Contents Camellia research A transcriptomic database of petal blight-resistant Camellia lutchuensis 47 Nikolai Kondratev1, Matthew Denton-Giles1,2, Cade D Fulton1, President’s message 6 Paul P Dijkwel1 -
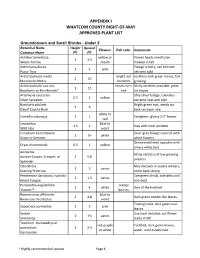
Whatcom County's Approved Plant List
APPENDIX I WHATCOM COUNTY RIGHT-OF-WAY APPROVED PLANT LIST Groundcovers and Small Shrubs - Under 2' Botanical Name Height Spread Flowers Fall color Comments Common Name (ft) (ft) Achillea tomentosa yellow or Flower heads need to be 1 3-5 Wooly Yarrow cream mowed in fall Antennaria dioica Foliage is furry, can tolerate 1 2 pink Pussy Toes extreme cold Arctostaphylos media bright red Leathery dark green leaves, fast 2 10 Manzanita Media branches growing Arctostaphylos uva ursi leaves turn Many varieties available, great 1 15 Bearberry or Kinnikinnick* red for slopes Artemesia caucasica Silky silver foliage, tolerates 0.5 2 yellow Silver Spreader extreme heat and cold Baccharis pilularis Bright green mat, needs cut 2 6 Dwarf Coyote Bush back once per year white to Camellia sasanqua 1 1 Evergreen, glossy 2-3" leaves red Ceanothus blue to 1.5 2 Stay with local varieties Wild Lilac violet Cerastium tomentosum Silver gray foliage covered with 1 5+ white Snow-in-Summer white flowers Ornamental seed capsules with Dryas drummondii 0.5 1 yellow silvery white tails Juniperus Many varieties of low growing Juniper-Carpet, Creeper, or 2 5-8 junipers Spreader Oenothera May die back in severe winters, 1 2 varies Evening Primrose come back strong Penstemon davidsonii, rupicola Evergreen shrub, tolerates cold 2 1.5 varies Beard Tongue and heat Pyracantha augustifolia orange 1 4 white One of the hardiest 'Gnome'* berries Rosmariunus officinalis blue to 2 4-8 Dark green needle-like leaves Rosemary 'Prostratus' violet Trailing habit, dark green oval Saponaria ocymoides 1 3 pink leaves Sedum Use local varieties, cut flower 2 1-5 varies Stonecrop stalks in fall Teucrium chamaedrys or red-purple Toothed, dark green leaves, postratium 1 2-3 or white water until established Germander * Highly-recommended species Page 1 Thymus white to 2 1-2 Many varieties Thyme purple Shrubs Botanical Name Height Spread Flowers Fall color Comments Common Name (ft) (ft) Arctostaphylos bright red Shiny dark green leaves turn 2-15 20 varies Manzanita berries maroon in fall. -

Not for Distribution
1 Efficacy of Tea in Human Health Isao Tomita* University of Shizuoka, Shizuoka, Japan Abstract Recent scientific findings on the effects of teaCamellia ( sinensis) on human health are reviewed. Some mechanistic explanations are discussed in relation to the special nature of (-)-epigallocatechin-3-gallate which works not only as an antioxidant but also as pro-oxidant. Though there are still some discrepan- cies between the results in animal models and those of epidemiological studies, the reasons will be uncovered in the near future. Keywords: antioxidant, chronic disease prevention, health effects, pro-oxidant, tea catechins 1.1 How the Physiological distribution which are soluble in hot water and show Effects Caused by Tea Drinking special physiological functions, such as Attracted Humans stimulation of the central nervous system. It is also well known that the tea leaves There are many legends whichfor told us to ex- contain a large amount of catechins (8–20% plain why the people in ancient China began of the dry weight) of which the major one is to drink tea. One of the stories told is about (-)-epigallocatechin-3-gallate (EGCG) (Fig.1.1). Wan Tu, the ancient Chinese emperor. He Tea catechins are oxidized to various was banished to a remote southern part of dimerization products, theaflavins, theasin- China (Yunnan province) due to his cruel ensins, and proantocyanidins and further to and tyrannizingNot governance. One day, he polymerization products, thearubigins, in the was sitting in the shade of a large bush in the process of tea preparation (Fig. 1.1). The taste area where Camellia sinensis grew and drank of tea is very unique: bitter, and astringent be- hot water. -

Processing and Chemical Constituents of Pu-Erh Tea: a Review
Food Research International 53 (2013) 608–618 Contents lists available at ScienceDirect Food Research International journal homepage: www.elsevier.com/locate/foodres Processing and chemical constituents of Pu-erh tea: A review Hai-peng Lv a,c, Ying-jun Zhang b,⁎, Zhi Lin c, Yue-rong Liang a,⁎⁎ a Zhejiang University Tea Research Institute, 866 Yuhangtang Road, Hangzhou 310058, PR China b State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, PR China c National Engineering Research Center for Tea Processing, Tea Research Institute, Chinese Academy of Agricultural Sciences (TRICAAS), 9 Meiling South Road, Hangzhou, Zhejiang 310008, PR China article info abstract Article history: Pu-erh tea is a unique microbial fermented tea produced from the sun-dried leaves of large-leaf tea species Received 15 October 2012 (Camellia sinensis (Linn.) var. assamica (Masters) Kitamura) in the Yunnan province of China. Pu-erh tea has Received in revised form 6 February 2013 become increasingly popular in Southeast Asia may be due to its multiple health benefits. The special sensory Accepted 21 February 2013 characteristics of Pu-erh tea arise from the multitudinous chemical changes and transformations of the chem- ical constituents of the sun-dried green tea leaves that occur during the post-fermentation process. Many Keywords: functional components have been isolated from Pu-erh tea and identified. In this paper, modern processing Pu-erh tea Chemical constituents techniques and their effects on the transformation of the chemical constituents and the major chemical com- Processing ponents of Pu-erh tea are reviewed and discussed.