Structural Asymmetry in a Conserved Signaling System That Regulates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of the Relatedness of Brucella Spp. and Ochrobactrum Anthropi and Description of Ochrobactrum Intermedium Sp
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Dadun, University of Navarra International Journal of Systematic Bacteriology (1 998), 48, 759-768 Printed in Great Britain Evaluation of the relatedness of Brucella spp. and Ochrobactrum anthropi and description of Ochrobactrum intermedium sp. nov., a new species with a closer relationship to Brucella SPP. Julian Velasco,’ Conchi Romero,’ lgnacio Lopez-Got%,’ Jose Leiva,2 Ramon Diaz1f2and lgnacio Moriydn’ Author for correspondence : Ignacio Moriyon. Tel : + 34 48 425600. Fax : + 34 48 425649. e-mail : [email protected] Departamento de The relatedness of Brucella spp. and Ochrobactrum anthropi was studied by M icrob io I og ia, Un ive rs id ad protein profiling, Western blot, immunoelectrophoresis and 16s rRNA analysis. de Navarra, Aptdo 1771 and Servicio de Microbiologia, Whole-cell and soluble proteins of brucellae and 0. anthropi showed Clinica Universitaria de serological cross-reactivities quantitatively and qualitatively more intense Navarraz, Pamplona, Spain than those existing with similar extracts of Agrobacterium spp. Numerical analysis of Western blot profiles of whole-cell extracts showed that 0. anthropi LMG 3301 was closer to Brucella spp. than to 0. anthropi LMG 3331T, a result not obtained by protein profiling. These differences were not observed by Western blot with soluble fractions, and immunoelectrophoretic analyses suggested that this was due to destruction of conformational epitopes in Western blot procedures with the subsequent simplification of antigenic profile. Analysis of the 165 rRNA sequences of strains previously used in the species definition confirmed that strain LMG 3301, and also LMG 3306, were closer to the brucellae, and that LMG 3331Twas in a separate cluster. -

Iron Transport Strategies of the Genus Burkholderia
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2015 Iron transport strategies of the genus Burkholderia Mathew, Anugraha Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-113412 Dissertation Published Version Originally published at: Mathew, Anugraha. Iron transport strategies of the genus Burkholderia. 2015, University of Zurich, Faculty of Science. Iron transport strategies of the genus Burkholderia Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr. sc. nat.) vorgelegt der Mathematisch-naturwissenschaftlichen Fakultät der Universität Zürich von Anugraha Mathew aus Indien Promotionskomitee Prof. Dr. Leo Eberl (Vorsitz) Prof. Dr. Jakob Pernthaler Dr. Aurelien carlier Zürich, 2015 2 Table of Contents Summary .............................................................................................................. 7 Zusammenfassung ................................................................................................ 9 Abbreviations ..................................................................................................... 11 Chapter 1: Introduction ....................................................................................... 14 1.1.Role and properties of iron in bacteria ...................................................................... 14 1.2.Iron transport mechanisms in bacteria ..................................................................... -

Caulobacter Crescentus
Biochemistry of the key spatial regulators MipZ and PopZ in Caulobacter crescentus Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) dem Fachbereich Biologie der Philipps-Universität Marburg vorgelegt von Yacine Refes aus Algier, Algerien Marburg, im November 2017 Vom Fachbereich Biologie der Philipps-Universität Marburg (Hochschulkennziffer: 1180) als Dissertation angenommen am: 22.01.2018 Erstgutachter: Prof. Dr. Martin Thanbichler Zweitgutachter: Prof. Dr. Torsten Waldminghaus Tag der mündlichen Prüfung am: 26.01.2018 Die Untersuchungen zur vorliegenden Arbeit wurden von November 2013 bis Juni 2017 am Max-Planck-Institut für terrestrische Mikrobiologie und an der Philipps Universität Marburg unter der Leitung von Prof. Dr. Martin Thanbichler durchgeführt. Publications Refes Y, He B, Corrales-Guerrero L, Steinchen W, Bange G, and Thanbichler M. Determinants of DNA binding by the bacterial cell division regulator MipZ. In preparation Corrales-Guerrero L, Refes Y, He B, Ramm B, Muecksch J, Steinchen W, Heimerl T, Knopp J, Bange G, Schwille P, and Thanbichler M. Regulation of cell division protein FtsZ by MipZ in Caulobacter crescentus. In preparation ...dans les champs de l'observation le hasard ne favorise que les esprits préparés - Louis Pasteur Abstract Bacteria are known to tightly control the spatial distribution of certain proteins by positioning them to distinct regions of the cell, particularly the cell poles. These regions represent important organizing platforms for several processes essential for bacterial survival and reproduction. The proteins localized at the cell poles are recruited to these positions by interaction with other polar proteins or protein complexes. The α-proteobacterium Caulobacter crescentus possesses a self- organizing polymeric polar matrix constituted of the scaffolding protein PopZ. -

Appendix a Bacteria
Appendix A Complete list of 594 pathogens identified in canines categorized by the following taxonomical groups: bacteria, ectoparasites, fungi, helminths, protozoa, rickettsia and viruses. Pathogens categorized as zoonotic/sapronotic/anthroponotic have been bolded; sapronoses are specifically denoted by a ❖. If the dog is involved in transmission, maintenance or detection of the pathogen it has been further underlined. Of these, if the pathogen is reported in dogs in Canada (Tier 1) it has been denoted by an *. If the pathogen is reported in Canada but canine-specific reports are lacking (Tier 2) it is marked with a C (see also Appendix C). Finally, if the pathogen has the potential to occur in Canada (Tier 3) it is marked by a D (see also Appendix D). Bacteria Brachyspira canis Enterococcus casseliflavus Acholeplasma laidlawii Brachyspira intermedia Enterococcus faecalis C Acinetobacter baumannii Brachyspira pilosicoli C Enterococcus faecium* Actinobacillus Brachyspira pulli Enterococcus gallinarum C C Brevibacterium spp. Enterococcus hirae actinomycetemcomitans D Actinobacillus lignieresii Brucella abortus Enterococcus malodoratus Actinomyces bovis Brucella canis* Enterococcus spp.* Actinomyces bowdenii Brucella suis Erysipelothrix rhusiopathiae C Actinomyces canis Burkholderia mallei Erysipelothrix tonsillarum Actinomyces catuli Burkholderia pseudomallei❖ serovar 7 Actinomyces coleocanis Campylobacter coli* Escherichia coli (EHEC, EPEC, Actinomyces hordeovulneris Campylobacter gracilis AIEC, UPEC, NTEC, Actinomyces hyovaginalis Campylobacter -
NVSL Reagent Manual
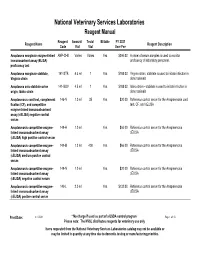
National Veterinary Services Laboratories Reagent Manual Reagent Amount/ Tests/ Billable FY 2021 Reagent Name Reagent Description Code Vial Vial User Fee Anaplasma marginale enzyme-linked ANP-CHK Varies VariesYes $394.00 A panel of serum samples is used to monitor immunosorbent assay (ELISA) proficiency of laboratory personnel. proficiency test Anaplasma marginale stabilate, 141-STB 4.5 ml 1Yes $188.00 Virginia strain, stabilate is used to initiate infection in Virginia strain donor animals Anaplasma ovis stabilate ovine 141-SOV 4.5 ml 1Yes $188.00 Idaho strain - stabilate is used to initate infection in origin, Idaho strain donor animals Anaplasmosis card test, complement 146-N 1.0 ml 35Yes $20.00 Reference control serum for the Anaplasmosis card fixation (CF), and competitive test, CF, and cELISA enzyme-linked immunoabsorbent assay (cELISA) negative control serum Anaplasmosis competitive enzyme- 149-H 1.0 mlYes $66.00 Reference control serum for the Anaplasmosis linked immunoabsorbent assay cELISA (cELISA) high positive control serum Anaplasmosis competitive enzyme- 149-M 1.0 ml 400Yes $66.00 Reference control serum for the Anaplasmosis linked immunoabsorbent assay cELISA (cELISA) medium positve control serum Anaplasmosis competitive enzyme- 149-N 1.0 mlYes $20.00 Reference control serum for the Anaplasmosis linked immunoabsorbent assay cELISA (cELISA) negative control serum Anaplasmosis competitive enzyme- 149-L 2.0 mlYes $132.00 Reference control serum for the Anaplasmosis linked immunoabsorbent assay cELISA (cELISA) positve control serum Print Date: 6/11/2021 * No charge if used as part of a USDA control program Page 1 of 58 Please note: The NVSL distributes reagents for veterinary use only Items requested from the National Veterinary Services Laboratories catalog may not be available or may be limited in quantity at any time due to domestic testing or manufacturing priorities. -

APUTS) Reporting Terminology and Codes Microbiology (V1.0
AUSTRALIAN PATHOLOGY UNITS AND TERMINOLOGY (APUTS) Reporting Terminology and Codes Microbiology (v1.0) 1 12/02/2013 APUTS Report Information Model - Urine Microbiology Page 1 of 1 Specimen Type Specimen Macro Time Glucose Bilirubin Ketones Specific Gravity pH Chemistry Protein Urobilinogen Nitrites Haemoglobin Leucocyte Esterases White blood cell count Red blood cells Cells Epithelial cells Bacteria Microscopy Parasites Microorganisms Yeasts Casts Crystals Other elements Antibacterial Activity No growth Mixed growth Urine MCS No significant growth Klebsiella sp. Bacteria ESBL Klebsiella pneumoniae Identification Virus Fungi Growth of >10^8 org/L 10^7 to 10^8 organism/L of mixed Range or number Colony Count growth of 3 organisms 19090-0 Culture Organism 1 630-4 LOINC >10^8 organisms/L LOINC Significant growth e.g. Ampicillin 18864-9 LOINC Antibiotics Susceptibility Method Released/suppressed None Organism 2 Organism 3 Organism 4 None Consistent with UTI Probable contamination Growth unlikely to be significant Comment Please submit a repeat specimen for testing if clinically indicated Catheter comments Sterile pyuria Notification to infection control and public health departments PUTS Urine Microbiology Information Model v1.mmap - 12/02/2013 - Mindjet 12/02/2013 APUTS Report Terminology and Codes - Microbiology - Urine Page 1 of 3 RCPA Pathology Units and Terminology Standardisation Project - Terminology for Reporting Pathology: Microbiology : Urine Microbiology Report v1 LOINC LOINC LOINC LOINC LOINC LOINC LOINC Urine Microbiology Report -

Regulation of Cell Fate Asymmetry in Caulobacter Crescentus by a Complex of Two Component Signaling Proteins
Regulation of Cell Fate Asymmetry in Caulobacter crescentus by a Complex of Two Component Signaling Proteins Christos G. Tsokos AUG 0 3 201 B.A. Biological Sciences Li0RA R ISF Cornell University, Ithaca, NY, 2004 ARCHIVES SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN BIOLOGY AT THE MASSACHUSETTS INSTITUTE OF TECHNOLOGY SEPTEMBER 2011 0 2011 Christos G. Tsokos. All rights reserved. The author hereby grants MIT permission to reproduce and distribute publicly paper and electronic copies of this thesis document in whole or in part. Signature of Author: _________G._______ Christos G. Tsokos Department of Biology June 14, 2011 Certified by: Michael T. Laub Associate Professor of Biology Thesis supervisor Accepted by: Alan D. Grossman Praecis Professor of Biology Regulation of Cell Fate Asymmetry in Caulobacter crescentus by a Complex of Two Component Signaling Proteins by Christos G. Tsokos Submitted to the Department of Biology on June 14, 2011 in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology at the Massachusetts Institute of Technology Abstract Cellular asymmetry is critical to the generation of complexity in both metazoans and many microbes. However, several molecular mechanisms responsible for translating asymmetry into differential cell fates remain unknown. Caulobacter crescentus provides an excellent model to study this process because every division is asymmetric. One daughter cell, the stalked cell, is sessile and commits immediately to S phase. The other daughter, the swarmer cell, is motile and locked in G1. Cellular differentiation requires asymmetric distribution or activation of regulatory factors. -

The Role of Caulobacter Cell Surface Structures in Colonization of the Air-Liquid Interface
bioRxiv preprint doi: https://doi.org/10.1101/524058; this version posted April 14, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. THE ROLE OF CAULOBACTER CELL SURFACE STRUCTURES IN COLONIZATION OF THE AIR-LIQUID INTERFACE ARETHA FIEBIG Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, IL [email protected] Abstract In aquatic environments, Caulobacter spp. are often present at the boundary between liquid and air known as the neuston. I report an approach to study temporal features of Caulobacter crescentus colonization and pellicle biofilm development at the air-liquid interface, and have defined the role of cell surface structures in this process. At this interface, C. crescentus initially forms a monolayer of cells bearing a surface adhesin known as the holdfast. When excised from the liquid surface, this monolayer strongly adheres to glass. The monolayer subsequently develops into a three-dimensional structure that is highly enriched in clusters of stalked cells known as rosettes. As this pellicle film matures, it becomes more cohesive and less adherent to a glass surface. A mutant strain lacking a flagellum does not efficiently reach the surface, and strains lacking type IV pili exhibit defects in organization of the three-dimensional pellicle. Strains unable to synthesize holdfast fail to accumulate at the boundary between air and liquid and do not form a pellicle. Phase contrast images support a model whereby the holdfast functions to trap C. -

A Novel and Conserved Dnaa-Related Protein That Targets The
bioRxiv preprint doi: https://doi.org/10.1101/712307; this version posted July 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. HdaB: a novel and conserved DnaA-related protein that targets the RIDA process to stimulate replication initiation Antonio Frandi and Justine Collier* Department of Fundamental Microbiology, Faculty of Biology and Medicine, University of Lausanne, Quartier UNIL/Sorge, Lausanne, CH 1015, Switzerland *Correspondance: E-mail: [email protected] Telephone: +41 21 692 5610 Fax: +41 21 692 5605 1 bioRxiv preprint doi: https://doi.org/10.1101/712307; this version posted July 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Exquisite control of the DnaA initiator is critical to ensure that bacteria initiate chromosome replication in a cell cycle-coordinated manner. In many bacteria, the DnaA-related and replisome-associated Hda/HdaA protein interacts with DnaA to trigger the regulatory inactivation of DnaA (RIDA) and prevent over-initiation events. In the C. crescentus Alphaproteobacterium, the RIDA process also targets DnaA for its rapid proteolysis by Lon. The impact of the RIDA process on adaptation of bacteria to changing environments remains unexplored. Here, we identify a novel and conserved DnaA-related protein, named HdaB, and show that homologs from three different Alphaproteobacteria can inhibit the RIDA process, leading to over-initiation and cell death when expressed in actively growing C. crescentus cells. -

Characterizing the Abcr/Vtlr System in the Rhizobiales
Characterizing the AbcR/VtlR system in the Rhizobiales Lauren Marie Sheehan Dissertation submitted to the faculty of the Virginia Polytechnic Institute and State University in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical and Veterinary Sciences Clayton C. Caswell, Chair Thomas J. Inzana Birgit Scharf Nammalwar Sriranganathan April 20, 2018 Blacksburg, Virginia Keywords: Brucella, LysR-type transcriptional regulators, small RNAs Characterizing the AbcR/VtlR system in the Rhizobiales Lauren Marie Sheehan Abstract Rhizobiales encompass a diverse group of microbes, ranging from free-living, soil- dwelling bacteria to disease-causing, intracellular pathogens. Although the lifestyle of these organisms vary, many genetic systems are well conserved. One system, named the AbcR/VtlR system, is found throughout rhizobiales, and even extends to bacteria in other orders within the Alphaproteobacteria. The AbcR sRNAs are an example of sibling sRNAs, where two copies of the abcR gene are typically present in the genome. The AbcRs are involved in the negative regulation of ABC-type transport systems, which are important components for nutrient acquisition. Although the AbcRs share several features amongst organisms, major differences can be found in their functional and regulatory redundancy, the targets they regulate and how they regulate them. Specifically, one major difference in the AbcRs lies in the nucleotide sequences utilized by the sRNAs to bind mRNA targets. In the present studies, the regulatory mechanisms of the AbcR sRNAs were further characterized in the mammalian pathogen Brucella abortus, and the full regulatory profiles of the AbcRs were defined in the plant pathogen Agrobacterium tumefaciens. As mentioned above, the AbcR sRNAs are important for the proper regulation of nutrient-acquiring transport systems in the Rhizobiales. -

The Development of Cellular Stalks in Bacteria
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central THE DEVELOPMENT OF CELLULAR STALKS IN BACTERIA JEAN M. SCHMIDT and R. Y. STANIER From the Department of Bacteriology and Immunology and the Electron Microscope Labora- tory, University of California, Berkeley ABSTRACT Extensive stalk elongation in Caulobacter and Asticcacaulis can bc obtained in a dcfincd medium by limiting thc concentration of phosphate. Caulobacter cells which wcre initiating stalk formation werc labeled with tritiatcd glucose. After rcmoval of exogenous tritiated material, the cells wcrc sul~jected to phosphate limitation whilc stalk elongation occurred. Thc location of tritiatcd material in the elongated stalks as dctected by radioautographic techniques allowed identification of the site of stalk development. Thc labeling pattern ob- tained was consistent with the hypothesis that the materials of the stalk are synthcsizcd at thc juncturc of the stalk with the cell. Complemcntary labcling experiments with Caulo- bacter and Asticcacaulis confirmed this result. In spheroplasts of C. crescentus preparcd by treatment with lysozyme, the stalks lost their normal rigid outline after several minutes of cxposurc to the enzyme, indicating that the rigid layer of the cell wall attacked by lysozyme is present in the stalk. In spheroplasts of growing cells induced with penicillin, the stalks did not appear to be affected, indicating that the stalk wall is a relatively inert, nongrowing structure. The morphogcnetic implications of these findings are discussed. INTRODUCTION The family Caulobacteraceae consists of rod- characteristically secreted at the pole of the cell. shaped or vibrioid bacteria, which can produce In Caulobacter, the holdfast consequently occurs filiform extensions of the cell, devoid of reproduc- around the terminal end of the stalk, but in tive function, known as stalks. -

Tese Adelina Margarida Parente
Defences of Helicobacter species against host antimicrobials Adelina Margarida Lima Pereira Rodrigues Parente Dissertation presented to obtain the Ph.D degree in Biochemistry Instituto de Tecnologia Química e Biológica António Xavier | Universidade Nova de Lisboa Oeiras, June, 2016 Defences of Helicobacter species against host antimicrobials Adelina Margarida Lima Pereira Rodrigues Parente Dissertation presented to obtain the Ph.D degree in Biochemistry Instituto de Tecnologia Química e Biológica António Xavier | Universidade Nova de Lisboa Oeiras, June 2016 From left to right: Mónica Oleastro (4th oponente), Gabriel Martins (3 rd opponent), Marta Justino (Co-supervisor) , Miguel Viveiros (2 nd opponent), Adelina Margarida Parente , Lígia Saraiva (supervisor) , Cecília Arraiano (president of the jury), and Maria do Céu Figueiredo (1 st opponent). nd 22 June 2016 Second edition, June 2016 Molecular Mechanisms of Pathogen Resistance Laboratory Instituto de Tecnologia Química e Biológica António Xavier Universidade Nova de Lisboa 2780-157 Portugal ii “Science knows no country, because knowledge belongs to humanity, and is the torch which illuminates the world” Louis Pasteur iii iv Acknowledgments Firstly, I would like to express my gratitude to the person that allowed me the opportunity to perform a PhD and who also contributed the most for my accomplishment of this thesis by constantly supporting me during these last years. I thus thank my supervisor, Dr. Lígia Saraiva , for her permanent availability whenever I needed guidance, for all the excellent ideas and advices related to my practical work and lastly for all the patience and enthusiasm! I also thank Dr. Lígia for her rigour and enormous help in the writing of this thesis.