Advances in Polymer Science
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2007 PMSE Fellow Ceremony
DIVISION OF POLYMERIC MATERIALS: SCIENCE & ENGINEERING 2007 PMSE Fellow Ceremony The American Chemical Society Division of Polymeric Materials: Science and Engineering (PMSE) has just completed its process to select a new class of Fellows for 2007, and the following people have been chosen: • James Crivello • Mohamed El-Aasser • James Stoffer • Wen-Li Wu They will be inducted as the eighth class of PMSE Fellows during the PMSE/POLY Awards Reception at the Chicago ACS National Meeting on Monday, March 26, 2007. PMSE is pleased to welcome this distinguished group of polymer scientists and engineers to the ranks of fellows. A short description of their work up to the point of the induction as a PMSE Fellow is on the following pages. http://www.pmsedivision.org DIVISION OF POLYMERIC MATERIALS: SCIENCE & ENGINEERING 2007 PMSE Fellow Induction Biographies 2007 PMSE Fellow James Crivello Rensselaer Polytechnic Institute Prof. James Crivello received his B.S. in chemistry from Aquinas College in Grand Rapids, Michigan in 1962 and his Ph.D. in organic chemistry from the University of Notre Dame in 1966. He joined the General Electric Corporate Research and Development Center in 1966 and was for several years a research project manger. His fields of activity include: organic nitrations, oxidations, and arylations, polyimides, silicones, and new photo- and thermal initiators for cationic and free radical polymerizations. In 1980, he was elected a Coolidge Fellow by the staff at GE Corporate Research and Development and spent the 1986-87 year as a visiting scientist at the University of Mainz with Prof. Helmut Ringsdorf in the Federal Republic of Germany. -

Liste Des Lauréats Français Du Prix Gay-Lussac Humboldt
Liste des lauréats français du prix Gay-Lussac Humboldt >Pierre AGOSTINI (2003), Physique atomique et moléculaire, CEA* Saclay > Ohio State University Josef DEUTSCHER (2005), Microbiologie, Institut National de la Recherche Agronomique - Jacques F. ARVIEUX (1994), Physique nucléaire, Université Paris 11 CNRS, Grignon Alain ASPECT (1999), Physique, Institut d’optique, Université Paris 11 – CNRS Georges DIDI-HUBERMAN (2006), Histoire de l’art, Ecole des Hautes Etudes en Sciences Didier ASTRUC (1988), Chimie, Université Bordeaux 1 Sociales Monique AUMAILLEY (1991), Biochimie cellulaire, Université Claude Bernard - Lyon 1 > Université Pierre-Henri DIXNEUF (1989), Chimie, Université Rennes 1 de Cologne Abdelhak DJOUADI (2007), Physique des particules, Université Paris 11 Claude BARDOS (1993), Mathématiques, Université Denis Diderot - Paris 7 Jean-François DUBREMETZ (1996), Biologie, Institut national de la santé et de la recherche Jean-Marie BASSET (1987), Chimie inorganique, CNRS**, Laboratoire de chimie médicale, Villeneuve d’Ascq CNRS - Université Montpellier 2 organométallique de surface, Lyon Magda ERICSON (1992), Physique nucléaire, Université Claude Bernard - Lyon I Henri C. BENOIT (1986), Chimie macromoléculaire, Université Louis Pasteur - Strasbourg 1 Gérard FEREY (2004), Chimie, Université de Versailles - Saint-Quentin Alain BENSOUSSAN (1983), Informatique, Institut national de recherche en informatique Jean-Marie FLAUD (1998), Chimie physique, Université Paris 11 et en automatique > Centre national d’études spatiales Christos FLYTZANIS -

Ivan Julian Dmochowski, Ph.D
Ivan Julian Dmochowski, Ph.D. Department of Chemistry, University of Pennsylvania 231 South 34 th Street, Philadelphia, PA 19104-6323 P: 215-898-6459; Email: [email protected] URL: http://dmochowskigroup.chem.upenn.edu/index.html Academic Appointments Professor of Chemistry, University of Pennsylvania, July, 2015 – Present Undergraduate Chair of Chemistry, University of Pennsylvania, Jan, 2015 – Present Associate Professor of Chemistry, University of Pennsylvania, 2010 – 2015 Assistant Professor of Chemistry, University of Pennsylvania, 2003 – 2010 Education California Institute of Technology, Pasadena, CA Helen Hay Whitney Postdoctoral Scholar in Biophysics, Sept. 2000 – Dec. 2002 California Institute of Technology, Pasadena, CA Ph.D. in Chemistry, May 2000, 1995-00 Johannes Gutenberg Universität, Mainz, Germany Research Fellow in Chemistry, 1994-95 Harvard College, Cambridge, MA A.B. in Chemistry, Magna cum Laude, 1990-94 Selected Honors 2016 Crano Award, Akron Section, American Chemical Society 2012 Awardee, McKnight Technological Innovations in Neuroscience 2011 Awardee, McGroddy Frontiers in Science, St. Joseph’s Univ. 2010 Invitee, National Academies Keck Futures Initiative Imaging Meeting 2007 Camille and Henry Dreyfus Teacher-Scholar Award 2005 NSF CAREER Award 2003 Camille and Henry Dreyfus New Faculty Award 2000 Herbert Newby McCoy Award, Caltech Chemistry Department 1990 United States Presidential Scholar Fellowships 2001-02 Helen Hay Whitney Postdoctoral Fellow 1999-00 N.I.H. Bioorganic/Bioinorganic Training Grant 1996-99 N.I.H. Biotechnology Training Grant Peer-Reviewed Publications 1 1. J.A. Rego, S. Kumar, I.J. Dmochowski, H. Ringsdorf, Synthesis of novel mixed tail triphenylene discotic liquid crystals - The search for higher order . Chem. Comm. (9) 1031- 1032, 1996. -
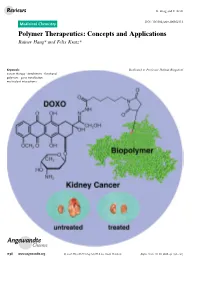
Polymer Therapeutics: Concepts and Applications Rainer Haag* and Felix Kratz*
Reviews R. Haag and F. Kratz DOI: 10.1002/anie.200502113 Medicinal Chemistry Polymer Therapeutics: Concepts and Applications Rainer Haag* and Felix Kratz* Keywords: Dedicated to Professor Helmut Ringsdorf cancer therapy · dendrimers · functional polymers · gene transfection · multivalent interactions Angewandte Chemie 1198 www.angewandte.org 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2006, 45, 1198 – 1215 Angewandte Polymer Therapeutics Chemie Polymer therapeutics encompass polymer–protein conjugates, drug– From the Contents polymer conjugates, and supramolecular drug-delivery systems. Numerous polymer–protein conjugates with improved stability and 1. Introduction 1199 pharmacokinetic properties have been developed, for example, by 2. Macromolecules as Drug- anchoring enzymes or biologically relevant proteins to polyethylene Delivery Systems: Biological glycol components (PEGylation). Several polymer–protein conjugates Rationale 1201 have received market approval, for example the PEGylated form of adenosine deaminase. Coupling low-molecular-weight anticancer 3. Approaches and Applications: “In Vivo Veritas” 1203 drugs to high-molecular-weight polymers through a cleavable linker is an effective method for improving the therapeutic index of clinically 4. Summary and Conclusions 1213 established agents, and the first candidates have been evaluated in clinical trials, including, N-(2-hydroxypropyl)methacrylamide conju- gates of doxorubicin, camptothecin, paclitaxel, and platinum(ii) complexes. Another class of polymer therapeutics are drug-delivery systems based on well-defined multivalent and dendritic polymers. is covalently linked to polymers such These include polyanionic polymers for the inhibition of virus as proteins, polysaccharides, or syn- thetic polymers. attachment, polycationic complexes with DNA or RNA (polyplexes), The coupling of drugs to macro- and dendritic core–shell architectures for the encapsulation of drugs. -

Ivan Julian Dmochowski, Ph.D
Ivan Julian Dmochowski, Ph.D. Department of Chemistry, University of Pennsylvania 231 South 34 th Street, Philadelphia, PA 19104-6323 URL: http://www.sas.upenn.edu/~ivandmo/ Phone: 215-898-6459; Fax: 215-898-2037 Email: [email protected] Academic Appointments Associate Professor of Chemistry, University of Pennsylvania, 2010 – Present Assistant Professor of Chemistry, University of Pennsylvania, 2003 – 2009 Education California Institute of Technology, Pasadena, CA Helen Hay Whitney Postdoctoral Scholar in Biophysics, Sept. 2000 – Dec. 2002 California Institute of Technology, Pasadena, CA Ph.D. in Chemistry, May 2000, 1995-00 Johannes Gutenberg Universität, Mainz, Germany Research Fellow in Chemistry, 1994-95 Harvard College, Cambridge, MA A.B. in Chemistry, Magna cum Laude, 1990-94 Selected Honors 2012 Awardee, McKnight Technological Innovations in Neuroscience 2011 Awardee, McGroddy Frontiers in Science, St. Joseph’s Univ. 2010 Invitee, National Academies Keck Futures Initiative Imaging Meeting 2007 Camille and Henry Dreyfus Teacher-Scholar Award 2005 NSF CAREER Award 2003 Camille and Henry Dreyfus New Faculty Award 2000 Herbert Newby McCoy Award, Caltech Chemistry Department 1990 United States Presidential Scholar Fellowships 2001-02 Helen Hay Whitney Postdoctoral Fellow 1999-00 N.I.H. Bioorganic/Bioinorganic Training Grant 1996-99 N.I.H. Biotechnology Training Grant Peer-Reviewed Publications: h-index = 21 (March, 2013) 1. J.A. Rego, S. Kumar, I.J. Dmochowski, H. Ringsdorf, Synthesis of novel mixed tail triphenylene discotic liquid crystals - The search for higher order. Chem. Comm. (9) 1031- 1032, 1996. 1 2. J.J. Wilker, I.J. Dmochowski, J.H. Dawson, J.R. Winkler, H.B. Gray, Substrates for rapid delivery of electrons and holes to buried active sites in proteins . -

Helmut Ringsdorf
Helmut Ringsdorf Department of Organic Chemistry, University of Mainz, Duesbergweg 10-14, 55099 Mainz, Germany, Phone +49 (0)6131 3922402, Fax +49 (0)6131 3923145, e-mail: [email protected] Helmut Ringsdorf was born in Gießen/Germany in 1929. He grew up in the Rhine Valley, studied Chemistry, Politics and Geology at the Universities in Frankfurt, Darmstadt, and Freiburg. In 1955 he entered Hermann Staudingers Lab as his very last student. After his Ph.D. in Freiburg in 1958, he was a Postdoctoral Fellow with C.G. Overberger and Herman F. Mark at the Polytechnic Institute of Brooklyn (1960-1962). He taught Polymer Science and Organic Chemistry at the Universities Marburg (1962-1970) and Mainz (1971-1994). He still has the good fortune to be with young scientists as Adjunct Professor at the School of Pharmacy, University Cardiff/U.K. and at the Jilin University, Changchun/China. His awards include the H. Staudinger (Germany) as well as the H. Mark (Austria) Award, Polymer Science Award (USA). Honorary doctors from four Universities in four countries, the Friendship Award of the Chinese Government, member of Academies in Germany (Mainz, Düsseldorf), Russia (Moscow), and Italy (Catania). Based on the concept of molecular engineering of functional polymers and molecular assemblies the research interests of the group in Mainz were centred around the attempt to bridge the gap between Materials Science and Life Science as summarized in the following figure. Main research themes: Molecular architecture of functional liquid crystalline polymers, polymerization in oriented systems (micelles, multicompartment micelles, liposomes, monolayers, multilayers), properties of functional supramolecular systems, attempts to mimic biomembrane processes (enzyme functions, protein docking) and polymer therapeutics, e.g. -

LIQUID CRYSTAL NEWSLIQUID NEWS $(%(&' "!!#$(%(&'"!!# GW Gray Medal for 2006 Professor Heino Finkelmann
LIQUID CRYSTAL NEWSLIQUID NEWS $(%(&' "!!#$(%(&'"!!# GW Gray Medal for 2006 Professor Heino Finkelmann as originally intended in Krakow. Since these days, we have met very frequently, for example, when I was privileged to lecture in Professor Finkelmann’s Institute and at many other scientific meetings not least of which was the Twelfth International Liquid Crystal Conference held in 1988 in Freiburg itself and chaired and organised by Professor Finkelmann. My wife and I well remember enjoying a glass of wine with Heino and his wife and two boys at their home after the closing ceremonies were complete and people like conference chairmen are at last allowed to relax. I have put the material in the above paragraph early in this article about Heino Finkelmann, because I wanted to stress that his very great contributions to the subject of liquid crystals have been not only in the area of scientific research, but also, by his readiness to travel far and wide to Heino Finkelmann was born in Gronau in Lower give lectures, by spreading the word on the subject, by Saxony in 1945. After school, his studies at the Scientific serving on the editorial boards of relevant journals, by Technical Academy at Isny were in the field of chemical organising meetings and conferences such as the 12th engineering and on the way to qualifying in 1969, he spent ILCC, by serving as he has twice done on the Board of time with Unilever Research and with British Petroleum in Directors of the International Liquid Crystal Society and by Hamburg. He then studied Chemistry at the Technical acting as Chairman of the German Liquid Crystal Society University of Berlin, and it was here that he first began as he did for many years. -

Cornell Chemistry November 1995 Number 64
Cornell Chemistry November 1995 Number 64 The Chairman's Notebook Science on the Silver Screen ne Friday morning last May at the American Society for Mass Spectrometry's annual meeting in Atlanta, nearly 900 scientists O dragged themselves out of bed for a plenary lecture at 8:15 a.m. Miles O'Brien, the keynote speaker, is the Cable News Network's lone science correspondent. Like many journalists who cover technological stories, O'Brien has no formal training in either mathematics or science. Once he discovered the secret to a successful story, however, those very news opportunities other reporters long ago eschewed quickly became his full-time job at CNN. His lecture was entitled 'The Geek Factor: ence that day in Atlanta, objecting to public with the excitement and passion Why Network Television Doesn't Cover O'Brien's characterization of the average only a top researcher or great teacher can Science." When conference organizers "Joe Six Pack" viewer, suggested that convey. marveled at the extraordinary turnout, CNN might have seriously underestimated O'Brien's lecture in Atlanta was full of O'Brien wryly observed that if you call its audience. Not so, O'Brien roared back. surprises. He showed videotapes of scientists by a derogatory name they will Whether they prefer cabernet, chardonnay, technology stories recently covered by always show up to find out why. Appar- Chivas, or Coors, viewers want to see CNN, some probing the underlying issues ently it goes with being an experimentalist. scientific stories on television with a clear, in depth over several minutes, which He explained that three ingredients are colorful message. -

Weck CV Feb 2021
Dr. Marcus Weck Curriculum Vitae MARCUS WECK Professor (212) 992-7968 (Voice) New York University (212) 995-4895 (Fax) Molecular Design Institute and Department of Chemistry e-mail: [email protected] New York, NY 10003-6688 http://weckresearch.com/home EDUCATIONAL BACKGROUND 1998 – 2000 Harvard University – Postdoctoral Fellow, Chemistry 1995 – 1998 California Institute of Technology – Ph.D., Chemistry 1988 – 1994 University of Mainz – M.Sc. (Diploma), Chemistry 1991 – 1992 University of California, Irvine – Exchange Student, Chemistry RESEARCH AND PROFESSIONAL EXPERIENCE 2007 – present New York University New York, NY Molecular Design Institute and Department of Chemistry · Director, NYU MRSEC 2017 – 2021 · Professor 2009 – present · Associate Director, Molecular Design Institute 2007 – present · Associate Professor 2007 – 2009 2000 – 2007 Georgia Institute of Technology Atlanta, GA School of Chemistry and Biochemistry · Associate Professor 2006 – 2007 · Assistant Professor 2000 – 2006 1998 – 2000 Harvard University Cambridge, MA Research Advisor: Prof. George M. Whitesides · Postdoctoral Research. Mimicking biological systems using mesoscale self- assembly. 1995 – 1998 California Institute of Technology Pasadena, CA Research Advisor: Prof. Robert H. Grubbs · Ph.D. Research. Olefin metathesis for the synthesis of supramolecular structures. 1993 – 1994 University of Mainz Mainz, Germany Research Advisor: Prof. Helmut Ringsdorf · Master’s Research. Molecular recognition at the air-water interface, in water and on solid supports. 1992 Max Planck Institute of Polymer Research Mainz, Germany · Summer Internship. Synthesis of phthalocyanines containing germanium and silicon. 1991 – 1992 University of California, Irvine Irvine, CA Research Advisor: Prof. Fraser Armstrong · Undergraduate Research. Voltammetric characterization of an iron-sulfur [4Fe-4S] cluster in ferredoxin III from Desulfovibrio africanus. CV-1 Dr. -

Advances in Polymer Science
262 Advances in Polymer Science Editorial Board: A. Abe, Tokyo, Japan A.-C. Albertsson, Stockholm, Sweden G.W. Coates, Ithaca, NY, USA J. Genzer, Raleigh, NC, USA S. Kobayashi, Kyoto, Japan K.-S. Lee, Daejeon, South Korea L. Leibler, Paris, France T.E. Long, Blacksburg, VA, USA M. Mo¨ller, Aachen, Germany O. Okay, Istanbul, Turkey B.Z. Tang, Hong Kong, China E.M. Terentjev, Cambridge, UK M.J. Vicent, Valencia, Spain B. Voit, Dresden, Germany U. Wiesner, Ithaca, NY, USA X. Zhang, Beijing, China For further volumes: http://www.springer.com/series/12 Aims and Scope The series Advances in Polymer Science presents critical reviews of the present and future trends in polymer and biopolymer science. It covers all areas of research in polymer and biopolymer science including chemistry, physical chemistry, physics, material science. The thematic volumes are addressed to scientists, whether at universities or in industry, who wish to keep abreast of the important advances in the covered topics. Advances in Polymer Science enjoys a longstanding tradition and good reputa- tion in its community. Each volume is dedicated to a current topic, and each review critically surveys one aspect of that topic, to place it within the context of the volume. The volumes typically summarize the significant developments of the last 5 to 10 years and discuss them critically, presenting selected examples, explaining and illustrating the important principles, and bringing together many important references of primary literature. On that basis, future research directions in the area can be discussed. Advances in Polymer Science volumes thus are important refer- ences for every polymer scientist, as well as for other scientists interested in polymer science - as an introduction to a neighboring field, or as a compilation of detailed information for the specialist. -

63Rd Otto M. Smith Banquet
A L PHA D ELTA CHAPTER O F PHI LAMBDA UPSILON presents the 63RD OTTO M. SMITH BANQUET with guest lecturer D R. C HRISTOPHER CUMMINS Professor of Chemistry Massachusetts Institute of Technology MARCH 7 , 2014 EVENING PROGRAM WELCOME MASTER OF CEREMONIES: RANGIKA HIKKADUWA DR. FRANK D . BLUM RECOGNITION OF DR. OTTO M. SMITH PRESENTATION OF AWARDS RECOGNITION OF GRADUATE ACCOMPLISHMENTS 0 .C . DERMER SCHOLARSHIP PRESENTED BY: DR. KENNETH D . BERLIN LIONEL M. RAFF SCHOLARSHIP PRESENTED BY: DR. FRANK BLUM PRESENTATION BY DR. CHRISTOPHER CUMMINS 20 1 3 .. 20 1 4 OFFICERS PRESIDENT RANGIKA HIKKADUUA VICE PRESIDENT NUWAN PERERA TREASURER TRAVIS JAMES COUNCILORS DR. BARRY LAVINE DR. ANDREW MORT 20 1 4 .. 20 1 5 O FFICERS PRESIDENT TBA VICE PRESIDENT TBA TREASURER TBA SECRETARY TBA SOCIAL CHAIR TBA DR. CHRISTOPHER CUMMINS Christopher "Kit" Colin Cummins was born in Boston, Massachusetts, and grew up also in New Orleans, Louisiana and then in Bloomington, Minnesota. As a young man his interests included reading, raising butterflies, farming, fi shing, canoe expeditions, sailing, gymnastics, and diving. In college, Kit benefited from fonnative undergraduate research experiences carried out sequentially in the laboratories of Professors Susan E Kegley, James P Collman, and Peter T Wolczanski, respectively of Middlebury College, Stanford University and Cornell University. He graduated from the latter institution with an AB degree in 1989. Following this he undertook inorganic chemistry graduate studies under the direction of Professor Richard R. Schrock at the Massachusetts Institute of Technology, from which he obtained his PhD degree in 1993 with a thesis entitled "Synthetic Investigations Featuring Amidometallic Complexes". -

Encyclopedia of Polymeric Nanomaterials
Encyclopedia of Polymeric Nanomaterials Shiro Kobayashi • Klaus Mullen€ Editors Encyclopedia of Polymeric Nanomaterials With 2021 Figures and 146 Tables Editors Shiro Kobayashi Klaus Mullen€ Center for Fiber and Textile Science Max Planck Institute Kyoto Institute of Technology for Polymer Research Matsugasaki, Sakyo-ku, Kyoto, Japan Mainz, Germany ISBN 978-3-642-29647-5 ISBN 978-3-642-29648-2 (eBook) ISBN 978-3-642-29649-9 (print and electronic bundle) DOI 10.1007/978-3-642-29648-2 Library of Congress Control Number: 2015941019 Springer Heidelberg New York Dordrecht London # Springer-Verlag Berlin Heidelberg 2015 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. The publisher, the authors and the editors are safe to assume that the advice and information in this book are believed to be true and accurate at the date of publication. Neither the publisher nor the authors or the editors give a warranty, express or implied, with respect to the material contained herein or for any errors or omissions that may have been made.