The Complete Chloroplast Genome Sequence of Mahonia Bealei
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Designations for Individual Genomes and Chromosomes in Gossypium WANG Kunbo1*, WENDEL Jonathan F.2 and HUA Jinping3
WANG et al. Journal of Cotton Research (2018) 1:3 Journal of Cotton Research https://doi.org/10.1186/s42397-018-0002-1 REVIEW Open Access Designations for individual genomes and chromosomes in Gossypium WANG Kunbo1*, WENDEL Jonathan F.2 and HUA Jinping3 Abstract Gossypium, as the one of the biggest genera, the most diversity, and the highest economic value in field crops, is assuming an increasingly important role in studies on plant taxonomy, polyploidization, phylogeny, cytogenetics, and genomics. Here we update and provide a brief summary of the emerging picture of species relationships and diversification, and a set of the designations for individual genomes and chromosomes in Gossypium. This cytogenetic and genomic nomenclature will facilitate comparative studies worldwide, which range from basic taxonomic exploration to breeding and germplasm introgression. Keywords: Nomenclature, Individual genome, Individual chromosome, Gossypium Because of its diversity and economic significance, the cotton diversity, from consisting of only a single species (F gen- genus (Gossypium) has been subjected to decades of taxo- ome) to larger genome groups containing more than a nomic, cytogenetic, and phylogenetic analyses. Accordingly, dozen species each (D, K). The important allopolyploid a reasonably well-documented phylogenetic and taxonomic clade, which includes G. hirsutum and G. barbadense,con- understanding has developed, as recently summarized tains 7 species, including two described only in the last (Wendel and Grover 2015). Work published since that time 10 years (G.ekmanianum,G.stephensii) (Krapovickas and also supports the emerging picture of species relationships Seijo 2008; Gallagher et al. 2017). and diversification (Grover et al. 2015a, 2015b;Chenetal. Many Gossypium species are taxonomically well- 2016; Gallagher et al. -

Largest Species, Citheronia Splendens Sinaloensis (Hoffmann) and Ea Cles Oslari Rothschild, Is Poorly Known
Journal of the Lepidopterists' Society 40(4), 1986, 264- 270 BIOLOGY AND IMMATURE STAGES OF CITHERONIA SPLENDENS SINALOENSIS AND EACLES OSLARI IN ARIZONA (SATURNIIDAE) PAUL M. TUSKES 7900 Cambridge IllD, Houston, Texas 77054 ABSTRACT. Citheronia splendens sinaloensis and Eacles oslari occur in Cochise, Pima, and Santa Cruz counties in southern Arizona. Both species have one generation per year. The flight season of E. oslari extends from early June to mid-August, and the larval host plants include Quercus species. The flight season of C. splendens extends from July to mid-August, and the larval host plants include wild cotton, manzanita, and New Mexico evergreen sumac. The immature stages are described for the first time. The citheroniine fauna of Arizona is unique in that all seven species are primarily of Mexican origin (Tuskes 1985). The biology of the two largest species, Citheronia splendens sinaloensis (Hoffmann) and Ea cles oslari Rothschild, is poorly known. Ferguson (1971) illustrated the adults, summarized existing information, and indicated that their im mature stages were undescribed. The purpose of this paper is to de scribe the immature stages of both species and to present additional biological and distributional information. Citheronia splendens sinaloensis (Figs. 1-4) Citheronia splendens sinaloensis is the only member of the genus presently known to occur in Arizona. Citheronia mexicana G. & R. occurs just south of Arizona, in Sonora, Mexico. Although reported from Arizona before the turn of the century, there are no recent United States records. Citheronia regalis (F.) and C. sepulcralis (Druce) are common in the eastern or central United States but do not occur farther west than central Texas. -

Population Structure and Genetic Diversity of the Boll Weevil (Coleoptera: Curculionidae) on Gossypium in North America Adam P
Entomology Publications Entomology 2012 Population Structure and Genetic Diversity of the Boll Weevil (Coleoptera: Curculionidae) on Gossypium in North America Adam P. Kuester Iowa State University, [email protected] Robert W. Jones Centro Universitario Queretaro Thomas W. Sappington Iowa State University, [email protected] Kyung Seok Kim Seoul National University NFoorllomwa nthi Bs. Bandarr additional works at: https://lib.dr.iastate.edu/ent_pubs UnitPeda Srta oftes theDepaArtmgricenulturt of Agalric Sulctuierence Commons, Agriculture Commons, Agronomy and Crop Sciences Commons, Biology Commons, Entomology Commons, Genetics Commons, and the See next page for additional authors Systems Biology Commons The ompc lete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ ent_pubs/198. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Entomology at Iowa State University Digital Repository. It has been accepted for inclusion in Entomology Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Population Structure and Genetic Diversity of the Boll Weevil (Coleoptera: Curculionidae) on Gossypium in North America Abstract Although the boll weevil, Anthonomus grandis grandis Boheman (Coleoptera: Curculionidae), is a devastating pest in the United States and Mexico, its population structure and genetic diversity -

Normas Para Confecção Da Versão
UNIVERSIDADE FEDERAL DE UBERLÂNDIA INSTITUTO DE GENÉTICA E BIOQUÍMICA PÓS-GRADUAÇÃO EM GENÉTICA E BIOQUÍMICA Poliploidia e variações reprodutivas em Bombacoideae (Malvaceae): distribuição geográfica, filogeografia e tamanho do genoma Aluna: Rafaela Cabral Marinho Orientadora: Profª. Drª. Ana Maria Bonetti Co-orientador: Prof. Dr. Paulo Eugênio Alves Macedo de Oliveira UBERLÂNDIA - MG 2017 UNIVERSIDADE FEDERAL DE UBERLÂNDIA INSTITUTO DE GENÉTICA E BIOQUÍMICA PÓS-GRADUAÇÃO EM GENÉTICA E BIOQUÍMICA Poliploidia e variações reprodutivas em Bombacoideae (Malvaceae): distribuição geográfica, filogeografia e tamanho do genoma Aluna: Rafaela Cabral Marinho Orientadora: Profª. Drª. Ana Maria Bonetti Co-orientador: Prof. Dr. Paulo Eugênio Alves Macedo de Oliveira Tese apresentada à Universidade Federal de Uberlândia como parte dos requisitos para obtenção do Título de Doutora em Genética e Bioquímica (Área Genética) UBERLÂNDIA – MG 2017 ii Dados Internacionais de Catalogação na Publicação (CIP) Sistema de Bibliotecas da UFU, MG, Brasil. M338p Marinho, Rafaela Cabral, 1988 2017 Poliploidia e variações reprodutivas em Bombacoideae (Malvaceae): distribuição geográfica, filogeografia e tamanho do genoma / Rafaela Cabral Marinho. - 2017. 100 f. : il. Orientadora: Ana Maria Bonetti. Coorientador: Paulo Eugênio Alves Macedo de Oliveira. Tese (doutorado) - Universidade Federal de Uberlândia, Programa de Pós-Graduação em Genética e Bioquímica. Disponível em: http://dx.doi.org/10.14393/ufu.di.2018.134 Inclui bibliografia. 1. Genética - Teses. 2. Malvaceae -

Yarra Yarra Group Inc (Incorporation No
Australian Plants Society Yarra Yarra Group Inc (Incorporation No. A0039676Y) Newsletter May 2018 May 3: Maree & Graham Goods : Gardening in the Wimmera Graham & Maree come from a farming and administration background. They are Life Members of the Wimmera Growers of Australian Plants of which they have been members for over 40 years. Their garden has been open to the public on several occasions for various charities. They ran a wholesale nursery for several years specialising in Eremophilas. Graham and Maree’s current project is growing plants, landscaping and overseeing the planting of the gardens surrounding the new building of their Church which was opened recently. They first became involved with Australia’s inland deserts in 2002 with an organisation called Desert Discovery as part of the Botany team. In 2008, Maree became the Botany Team Leader, while Graham was involved with the photography and identification of specimens. They have done volunteer work for Australian Wildlife Conservancy, Friends of the Great Victoria Desert, Victorian and Western Australian Herbariums in collecting, identifying and processing specimens. Graham and Maree are co-authors of the book, Birds and Plants of the Little Desert, a photographic guide and Maree a co-author of Australia’s Eremophilas Website: apsyarrayarra.org.au Facebook: facebook.com/APSYarraYarra Email: [email protected] | 1 APS Yarra Yarra Particulars APS YY General Meeting APS YY Garden Visits: Speakers: May 13, 2 pm, Bill Aitchison and Sue Guymer Garden, Donvale. 7-June Greg Moore Urban Greening 5-July Ryan Phillips Animal Interactions Natural bush garden with ponds,stream and large dam merging to adjacent bush. -

Explorations in Ethnobiology: the Legacy of Amadeo Rea
Explorations in Ethnobiology: The Legacy of Amadeo Rea Edited by Marsha Quinlan and Dana Lepofsky Explorations in Ethnobiology: The Legacy of Amadeo Rea Edited by Marsha Quinlan and Dana Lepofsky Copyright 2013 ISBN-10: 0988733013 ISBN-13: 978-0-9887330-1-5 Library of Congress Control Number: 2012956081 Society of Ethnobiology Department of Geography University of North Texas 1155 Union Circle #305279 Denton, TX 76203-5017 Cover photo: Amadeo Rea discussing bird taxonomy with Mountain Pima Griselda Coronado Galaviz of El Encinal, Sonora, Mexico, July 2001. Photograph by Dr. Robert L. Nagell, used with permission. Contents Preface to Explorations in Ethnobiology: The Legacy of Amadeo Rea . i Dana Lepofsky and Marsha Quinlan 1 . Diversity and its Destruction: Comments on the Chapters . .1 Amadeo M. Rea 2 . Amadeo M . Rea and Ethnobiology in Arizona: Biography of Influences and Early Contributions of a Pioneering Ethnobiologist . .11 R. Roy Johnson and Kenneth J. Kingsley 3 . Ten Principles of Ethnobiology: An Interview with Amadeo Rea . .44 Dana Lepofsky and Kevin Feeney 4 . What Shapes Cognition? Traditional Sciences and Modern International Science . .60 E.N. Anderson 5 . Pre-Columbian Agaves: Living Plants Linking an Ancient Past in Arizona . .101 Wendy C. Hodgson 6 . The Paleobiolinguistics of Domesticated Squash (Cucurbita spp .) . .132 Cecil H. Brown, Eike Luedeling, Søren Wichmann, and Patience Epps 7 . The Wild, the Domesticated, and the Coyote-Tainted: The Trickster and the Tricked in Hunter-Gatherer versus Farmer Folklore . .162 Gary Paul Nabhan 8 . “Dog” as Life-Form . .178 Eugene S. Hunn 9 . The Kasaga’yu: An Ethno-Ornithology of the Cattail-Eater Northern Paiute People of Western Nevada . -

Growing Australian Plants
ASSOqIATTON OF SOCIETIES FOR GROWING AUSTRALIAN PLANTS LETTER NO 11 :ISSN: 1488-1488 at Hibiscus species H. forsterii F.D. Wilson (Sect. Furcaria DC') Contaitrer srown guclerim (jueensland 6n May 2007 Images C' Harve) G. Harvey Hibiscus diversifolius winter bloom 2nd June 2007. lnage NL 1l p.l Welcome to Newsletter No I l The front page depicts H. forsterii from the Cape York region of Queensland. The image of H. diversifolius blooms at the foot of the page contrast sharply with the purple flowers obtained in summer ; (see front page of Newsletter No 10). When the wirrter temperature is not quite so cold the off-white colour seen in the image, tends towards pale lemon and with still warmer weather becomes pink and finally red/purple in summer. It is being sold in Fairhill Native Plant Nursery as "Colour Magic". The so called'Norfolk Island Hibiscus" : Lagunaria patersonius (Andrews) G. Don subsp. patersonius, has appeared on a Norfolk Island postage stamp as illustrated bottom right on the front page. Winter arrived with a vengeance in the first week of June following a very mild autumn. Many Hibiscus came into bud early and H. heterophyllus are now in full bloom on the nofthern end of the Sunshine Coast. Dion Harrison reports some early blooms from the Mount Crosby Cliffs and Kenmore near Brisbane. At Buderim our rainfall for June was 211mm, our average for that month being 14lmm. Unfortunately this coastal rain hasn't penetrated inland to much extent, where it is much needed in the dam catchment areas. H. -

Vascular Plant and Vertebrate Inventory of Fort Bowie National Historic Site Vascular Plant and Vertebrate Inventory of Fort Bowie National Historic Site
Powell, Schmidt, Halvorson In Cooperation with the University of Arizona, School of Natural Resources Vascular Plant and Vertebrate Inventory of Fort Bowie National Historic Site Vascular Plant and Vertebrate Inventory of Fort Bowie National Historic Site Plant and Vertebrate Vascular U.S. Geological Survey Southwest Biological Science Center 2255 N. Gemini Drive Flagstaff, AZ 86001 Open-File Report 20 Southwest Biological Science Center Open-File Report 2005-1167 February 2007 05-1 U.S. Department of the Interior 167 U.S. Geological Survey National Park Service In cooperation with the University of Arizona, School of Natural Resources Vascular Plant and Vertebrate Inventory of Fort Bowie National Historic Site By Brian F. Powell, Cecilia A. Schmidt , and William L. Halvorson Open-File Report 2005-1167 December 2006 USGS Southwest Biological Science Center Sonoran Desert Research Station University of Arizona U.S. Department of the Interior School of Natural Resources U.S. Geological Survey 125 Biological Sciences East National Park Service Tucson, Arizona 85721 U.S. Department of the Interior DIRK KEMPTHORNE, Secretary U.S. Geological Survey Mark Myers, Director U.S. Geological Survey, Reston, Virginia: 2006 For product and ordering information: World Wide Web: http://www.usgs.gov/pubprod Telephone: 1-888-ASK-USGS For more information on the USGS-the Federal source for science about the Earth, its natural and living resources, natural hazards, and the environment: World Wide Web:http://www.usgs.gov Telephone: 1-888-ASK-USGS Suggested Citation Powell, B. F, C. A. Schmidt, and W. L. Halvorson. 2006. Vascular Plant and Vertebrate Inventory of Fort Bowie National Historic Site. -

November 2018 Serving the Mountain Empire Communities of Canelo, Elgin, Patagonia and Sonoita Vol
NOVEMBER 2018 SERVING THE MOUNTAIN EMPIRE COMMUNITIES OF CANELO, ELGIN, PATAGONIA AND SONOITA VOL. 8, ISSUE 9 By Aisha Sander The 30th Patagonia Fall Festival faced some unforeseen A Rainy Fall Festival challenges due to unfavorable weather conditions as well as discontent expressed by some of the participants with changes in the structure of the event. This year the event was outsourced to a professional event management company, South- ern Arizona Arts & Cultural Alliance (SAACA). For the past 29 years the Fall festival has been man- aged and put on by locals, volunteers and most recently by Sky Island Tourist Association (SITA). A SAACA representative said they were very excited to work at the Fall Festival, and that it was a perfect fit, as they specialize in creating art experiences. Their goals were to keep the artists happy and to give them opportunities for business and marketing. They also hoped to provide overall support to the town and the business community. As an organization that has primarily worked in the urban areas of Arizona, SAACA was excited to branch out to a rural area and merge the best of both worlds for the Fall Festival. SAACA intends to reinvest the funds generated from the festival by giving a portion to the town and retaining a portion for their own arts programming around Arizona. There were some significant changes with the new management. The first change was to remove Photo by Aisha Sander Friday as a full day for vendors, limiting it to a com- Two young boys enjoy the chance to play in the mud puddles in the Patagonia Park at the Fall Festival munity kick-off celebration. -

Antisense Suppression of a (+)-D-Cadinene Synthase Gene In
Antisense Suppression of a (1)-d-Cadinene Synthase Gene in Cotton Prevents the Induction of This Defense Response Gene during Bacterial Blight Infection But Not Its Constitutive Expression1[w] Belinda J. Townsend2, Andrew Poole, Christopher J. Blake, and Danny J. Llewellyn* Commonwealth Scientific and Industrial Research Organisation-Plant Industry, Canberra, Australian Capital Territory 2601, Australia (B.J.T., A.P., D.J.L.); and Department of Biochemistry and Molecular Biology (B.J.T.) and Research School of Chemistry (C.J.B.), Australian National University, Canberra, Australian Capital Territory 0200, Australia In cotton (Gossypium hirsutum) the enzyme (1)-d-cadinene synthase (CDNS) catalyzes the first committed step in the biosynthesis of cadinane-type sesquiterpenes, such as gossypol, that provide constitutive and inducible protection against pests and diseases. A cotton cDNA clone encoding CDNS (cdn1-C4) was isolated from developing embryos and functionally characterized. Southern analysis showed that CDNS genes belong to a large multigene family, of which five genomic clones were studied, including three pseudogenes and one gene that may represent another subfamily of CDNS. CDNS expression was shown to be induced in cotton infected with either the bacterial blight or verticillium wilt pathogens. Constructs for the constitutive or seed-specific antisense suppression of cdn1-C4 were introduced into cotton by Agrobacterium-mediated transformation. Gossypol levels were not reduced in the seeds of transformants with either construct, nor was the induction of CDNS expression affected in stems of the constitutive antisense plants infected with Verticillium dahliae Kleb. However, the induction of CDNS mRNA and protein in response to bacterial blight infection of cotyledons was completely blocked in the constitutive antisense plants. -
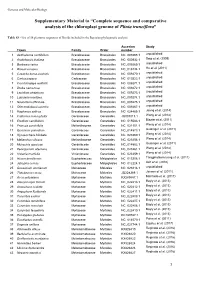
Complete Sequence and Comparative Analysis of the Chloroplast Genome of Plinia Trunciflora”
Genetics and Molecular Biology Supplementary Material to “Complete sequence and comparative analysis of the chloroplast genome of Plinia trunciflora” Table S3 - List of 56 plastome sequences of Rosids included in the Bayesian phylogenetic analysis. Accesion Study Taxon Family Order number 1 Aethionema cordifolium Brassicaceae Brassicales NC_009265.1 unpublished 2 Arabidopsis thaliana Brassicaceae Brassicales NC_000932.1 Sato et al. (1999) 3 Barbarea verna Brassicaceae Brassicales NC_009269.1 unpublished 4 Brassica napus Brassicaceae Brassicales NC_016734.1 Hu et al. (2011) 5 Capsella bursa-pastoris Brassicaceae Brassicales NC_009270.1 unpublished 6 Carica papaya Caricaceae Brassicales NC_010323.1 unpublished 7 Crucihimalaya wallichii Brassicaceae Brassicales NC_009271.1 unpublished 8 Draba nemorosa Brassicaceae Brassicales NC_009272.1 unpublished 9 Lepidium virginicum Brassicaceae Brassicales NC_009273.1 unpublished 10 Lobularia maritima Brassicaceae Brassicales NC_009274.1 unpublished 11 Nasturtium officinale Brassicaceae Brassicales NC_009275.1 unpublished 12 Olimarabidopsis pumila Brassicaceae Brassicales NC_009267.1 unpublished 13 Raphanus sativus Brassicaceae Brassicales NC_024469.1 Jeong et al. (2014) 14 California macrophylla Geraniaceae Geraniales JQ031013.1 Weng et al. (2014) 15 Erodium carvifolium Geraniaceae Geraniales NC_015083.1 Blazier et al. (2011) 16 Francoa sonchifolia Melianthaceae Geraniales NC_021101.1 Weng et al. (2014) 17 Geranium palmatum Geraniaceae Geraniales NC_014573.1 Guisinger et al. (2011) 18 Hypseocharis bilobate Geraniaceae Geraniales NC_023260.1 Weng et al. (2014) 19 Melianthus villosus Melianthaceae Geraniales NC_023256.1 Weng et al. (2014) 20 Monsonia speciose Geraniaceae Geraniales NC_014582.1 Guisinger et al. (2011) 21 Pelargonium alternans Geraniaceae Geraniales NC_023261.1 Weng et al. (2014) 22 Viviania marifolia Vivianiaceae Geraniales NC_023259.1 Weng et al. (2014) 23 Hevea brasiliensis Euphorbiaceae Malpighiales NC_015308.1 Tangphatsornruang et al. -

Downloaded from Genbank on That Full Plastid Genomes Are Not Sufficient to Reject Al- February 28, 2012
Ruhfel et al. BMC Evolutionary Biology 2014, 14:23 http://www.biomedcentral.com/1471-2148/14/23 RESEARCH ARTICLE Open Access From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes Brad R Ruhfel1*, Matthew A Gitzendanner2,3,4, Pamela S Soltis3,4, Douglas E Soltis2,3,4 and J Gordon Burleigh2,4 Abstract Background: Next-generation sequencing has provided a wealth of plastid genome sequence data from an increasingly diverse set of green plants (Viridiplantae). Although these data have helped resolve the phylogeny of numerous clades (e.g., green algae, angiosperms, and gymnosperms), their utility for inferring relationships across all green plants is uncertain. Viridiplantae originated 700-1500 million years ago and may comprise as many as 500,000 species. This clade represents a major source of photosynthetic carbon and contains an immense diversity of life forms, including some of the smallest and largest eukaryotes. Here we explore the limits and challenges of inferring a comprehensive green plant phylogeny from available complete or nearly complete plastid genome sequence data. Results: We assembled protein-coding sequence data for 78 genes from 360 diverse green plant taxa with complete or nearly complete plastid genome sequences available from GenBank. Phylogenetic analyses of the plastid data recovered well-supported backbone relationships and strong support for relationships that were not observed in previous analyses of major subclades within Viridiplantae. However, there also is evidence of systematic error in some analyses. In several instances we obtained strongly supported but conflicting topologies from analyses of nucleotides versus amino acid characters, and the considerable variation in GC content among lineages and within single genomes affected the phylogenetic placement of several taxa.