Microglia–Neuron Crosstalk in Obesity: Melodious Interaction Or Kiss of Death?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oligodendrocytes in Development, Myelin Generation and Beyond
cells Review Oligodendrocytes in Development, Myelin Generation and Beyond Sarah Kuhn y, Laura Gritti y, Daniel Crooks and Yvonne Dombrowski * Wellcome-Wolfson Institute for Experimental Medicine, Queen’s University Belfast, Belfast BT9 7BL, UK; [email protected] (S.K.); [email protected] (L.G.); [email protected] (D.C.) * Correspondence: [email protected]; Tel.: +0044-28-9097-6127 These authors contributed equally. y Received: 15 October 2019; Accepted: 7 November 2019; Published: 12 November 2019 Abstract: Oligodendrocytes are the myelinating cells of the central nervous system (CNS) that are generated from oligodendrocyte progenitor cells (OPC). OPC are distributed throughout the CNS and represent a pool of migratory and proliferative adult progenitor cells that can differentiate into oligodendrocytes. The central function of oligodendrocytes is to generate myelin, which is an extended membrane from the cell that wraps tightly around axons. Due to this energy consuming process and the associated high metabolic turnover oligodendrocytes are vulnerable to cytotoxic and excitotoxic factors. Oligodendrocyte pathology is therefore evident in a range of disorders including multiple sclerosis, schizophrenia and Alzheimer’s disease. Deceased oligodendrocytes can be replenished from the adult OPC pool and lost myelin can be regenerated during remyelination, which can prevent axonal degeneration and can restore function. Cell population studies have recently identified novel immunomodulatory functions of oligodendrocytes, the implications of which, e.g., for diseases with primary oligodendrocyte pathology, are not yet clear. Here, we review the journey of oligodendrocytes from the embryonic stage to their role in homeostasis and their fate in disease. We will also discuss the most common models used to study oligodendrocytes and describe newly discovered functions of oligodendrocytes. -

Build a Neuron
Build a Neuron Objectives: 1. To understand what a neuron is and what it does 2. To understand the anatomy of a neuron in relation to function This activity is great for ALL ages-even college students!! Materials: pipe cleaners (2 full size, 1 cut into 3 for each student) pony beads (6/student Introduction: Little kids: ask them where their brain is (I point to my head and torso areas till they shake their head yes) Talk about legos being the building blocks for a tower and relate that to neurons being the building blocks for your brain and that neurons send messages to other parts of your brain and to and from all your body parts. Give examples: touch from body to brain, movement from brain to body. Neurons are the building blocks of the brain that send and receive messages. Neurons come in all different shapes. Experiment: 1. First build soma by twisting a pipe cleaner into a circle 2. Then put a 2nd pipe cleaner through the circle and bend it over and twist the two strands together to make it look like a lollipop (axon) 3. take 3 shorter pipe cleaners attach to cell body to make dendrites 4. add 6 beads on the axon making sure there is space between beads for the electricity to “jump” between them to send the signal super fast. (myelin sheath) 5. Twist the end of the axon to make it look like 2 feet for the axon terminal. 6. Make a brain by having all of the neurons “talk” to each other (have each student hold their neuron because they’ll just throw them on a table for you to do it.) messages come in through the dendrites and if its a strong enough electrical change, then the cell body sends the Build a Neuron message down it’s axon where a neurotransmitter is released. -

Tanycytes of the Adult Hypothalamic Third Ventricle Include Distinct Populations of FGF-Responsive Neural Progenitors
ARTICLE Received 27 Nov 2012 | Accepted 23 May 2013 | Published 27 Jun 2013 DOI: 10.1038/ncomms3049 OPEN a-Tanycytes of the adult hypothalamic third ventricle include distinct populations of FGF-responsive neural progenitors S.C. Robins1,2,*, I. Stewart1,*, D.E McNay3,4,*, V. Taylor5, C. Giachino5, M. Goetz6, J. Ninkovic6, N. Briancon3,7, E. Maratos-Flier3, J.S Flier3,8, M.V Kokoeva2,3 & M. Placzek1 Emerging evidence suggests that new cells, including neurons, can be generated within the adult hypothalamus, suggesting the existence of a local neural stem/progenitor cell niche. Here, we identify a-tanycytes as key components of a hypothalamic niche in the adult mouse. Long-term lineage tracing in vivo using a GLAST::CreERT2 conditional driver indicates that a-tanycytes are self-renewing cells that constitutively give rise to new tanycytes, astrocytes and sparse numbers of neurons. In vitro studies demonstrate that a-tanycytes, but not b-tanycytes or parenchymal cells, are neurospherogenic. Distinct subpopulations of a-tanycytes exist, amongst which only GFAP-positive dorsal a2-tanycytes possess stem-like neurospherogenic activity. Fgf-10 and Fgf-18 are expressed specifically within ventral tanycyte subpopulations; a-tanycytes require fibroblast growth factor signalling to maintain their proliferation ex vivo and elevated fibroblast growth factor levels lead to enhanced proliferation of a-tanycytes in vivo. Our results suggest that a-tanycytes form the critical component of a hypothalamic stem cell niche, and that local fibroblast growth factor signalling governs their proliferation. 1 MRC Centre for Developmental and Biomedical Genetics and Department of Biomedical Science, University of Sheffield, Sheffield S10 2TN, UK. -

Neurons – Is a Basic Cell of the Nervous System. • Neurons Carry Nerve Messages, Or Impulses, from One Part of the Body to Another
Nervous System Nerves and Nerve Cells: Neurons – is a basic cell of the nervous system. • Neurons carry nerve messages, or impulses, from one part of the body to another. Structure of a Nerve Cell: A neuron has three basic parts: 1. Body – controls the cell’s growth 2. Axon – is a long thin fiber that carries impulses away from the cell body Myelin – is a fatty material that insulates the axon and increases the speed at which an impulse travels 3. Dendrites – are short, branching fibers that carry nerve impulses toward the cell body. • A nerve impulse begins when the dendrites are stimulated. The impulse travels along the dendrites to the cell body, and then away from the cell body on the axon. • The impulse must cross a synapse to a muscle or another neuron. Synapse – is the space between an axon and the structure with which the neuron communicates. Types of Nerve Cells: Sensory Neurons – pick up information about your external and internal environment from your sense organs and your body Motor Neurons – sends impulses to your muscles and glands, causing them to react Interneurons – are located only in the brain and spinal cord, pass impulses from one neuron to another The Central Nervous System: • The nervous system consists of two parts. Your brain and spinal cord make up one part, which is called the central nervous system. 1 • The peripheral nervous system, which is the other part, is made up of all the nerves that connect the brain and spinal cord to other parts of the body. The Brain: Brain ± a moist, spongy organ weighing about three pounds is made up of billions of neurons that control almost everything you do and experience. -

Endocannabinoids in Body Weight Control
pharmaceuticals Review Endocannabinoids in Body Weight Control Henrike Horn †, Beatrice Böhme †, Laura Dietrich and Marco Koch * Institute of Anatomy, Medical Faculty, University of Leipzig, 04103 Leipzig, Germany; [email protected] (H.H.); [email protected] (B.B.); [email protected] (L.D.) * Correspondence: [email protected]; Tel.: +49-341-97-22047 † These authors contributed equally. Received: 28 April 2018; Accepted: 28 May 2018; Published: 30 May 2018 Abstract: Maintenance of body weight is fundamental to maintain one’s health and to promote longevity. Nevertheless, it appears that the global obesity epidemic is still constantly increasing. Endocannabinoids (eCBs) are lipid messengers that are involved in overall body weight control by interfering with manifold central and peripheral regulatory circuits that orchestrate energy homeostasis. Initially, blocking of eCB signaling by first generation cannabinoid type 1 receptor (CB1) inverse agonists such as rimonabant revealed body weight-reducing effects in laboratory animals and men. Unfortunately, rimonabant also induced severe psychiatric side effects. At this point, it became clear that future cannabinoid research has to decipher more precisely the underlying central and peripheral mechanisms behind eCB-driven control of feeding behavior and whole body energy metabolism. Here, we will summarize the most recent advances in understanding how central eCBs interfere with circuits in the brain that control food intake and -

Myelin Biogenesis Is Associated with Pathological Ultrastructure That Is
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Myelin biogenesis is associated with pathological ultrastructure that 2 is resolved by microglia during development 3 4 5 Minou Djannatian1,2*, Ulrich Weikert3, Shima Safaiyan1,2, Christoph Wrede4, Cassandra 6 Deichsel1,2, Georg Kislinger1,2, Torben Ruhwedel3, Douglas S. Campbell5, Tjakko van Ham6, 7 Bettina Schmid2, Jan Hegermann4, Wiebke Möbius3, Martina Schifferer2,7, Mikael Simons1,2,7* 8 9 1Institute of Neuronal Cell Biology, Technical University Munich, 80802 Munich, Germany 10 2German Center for Neurodegenerative Diseases (DZNE), 81377 Munich, Germany 11 3Max-Planck Institute of Experimental Medicine, 37075 Göttingen, Germany 12 4Institute of Functional and Applied Anatomy, Research Core Unit Electron Microscopy, 13 Hannover Medical School, 30625, Hannover, Germany 14 5Department of Neuronal Remodeling, Graduate School of Pharmaceutical Sciences, Kyoto 15 University, Sakyo-ku, Kyoto 606-8501, Japan. 16 6Department of Clinical Genetics, Erasmus MC, University Medical Center Rotterdam, 3015 CN, 17 the Netherlands 18 7Munich Cluster of Systems Neurology (SyNergy), 81377 Munich, Germany 19 20 *Correspondence: [email protected] or [email protected] 21 Keywords 22 Myelination, degeneration, phagocytosis, microglia, oligodendrocytes, phosphatidylserine 23 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

View Software
TLR4-activated microglia have divergent effects on oligodendrocyte lineage cells DISSERTATION Presented in partial fulfillment of the requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University Evan Zachary Goldstein, BA Neuroscience Graduate Program The Ohio State University 2016 Dissertation Committee: Dr. Dana McTigue, Advisor Dr. Phillip Popovich Dr. Jonathan Godbout Dr. Courtney DeVries Copyright by Evan Zachary Goldstein 2016 i Abstract Myelin accelerates action potential conduction velocity and provides essential metabolic support for axons. Unfortunately, myelin and myelinating cells are often vulnerable to injury or disease, resulting in myelin damage, which in turn can lead to axon dysfunction, overt pathology and neurological impairment. Inflammation is a common component of CNS trauma and disease, and therefore an active inflammatory response is often considered deleterious to myelin health. While inflammation can certainly damage myelin, inflammatory processes also benefit oligodendrocyte (OL) lineage progression and myelin repair. Consistent with the divergent nature of inflammation, intraspinal toll-like receptor 4 (TLR4) activation, an innate immune pathway, kills OL lineage cells, but also initiates oligodendrogenesis. Soluble factors produced by TLR4-activated microglia can reproduce these effects in vitro, however the exact factors are unknown. To determine what microglial factors might contribute to TLR4-induced OL loss and oligodendrogenesis, mRNA of factors known to affect OL lineage cells was quantified in TLR4-activated microglia and spinal cords (chapter 2). Results indicate that TLR4-activated microglia transcribe numerous factors that induce OL loss, OL progenitor cell (OPC) proliferation and OPC differentiation. However, some factors upregulated after intraspinal TLR4 activation were not ii upregulated by microglia, suggesting that other cell types contribute to transcriptional changes in vivo. -
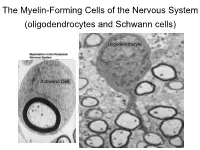
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Tecnicas Microscopicas
CAP 1: TÉCNICAS MICROSCÓPICAS TÉCNICAS MICROSCÓPICAS 11 Lic. Carlos R. Neira Montoya Lic. Eduardo Sedano Gelvet Lic. María Elena Vilcarromero V. El estudio de los tejidos tal como los observamos hoy en día no sería posible sin la ayuda de la histotecnología; esta disciplina se encarga del estudio de los métodos técnicas y procedimientos que permiten la transformación de un órgano en una película lo suficientemente transparente y contrastada que nos permite su observación a través del microscopio (Fig. 1-1). Para que esto ocurra se tiene que seguir una serie de pasos. Cada uno de estos pasos permite la observación de las características morfológicas del tejido que nos indica la normalidad o la alteración patológica; sin embargo en estudios mucho más minuciosos, estos pasos se harán en función de las estructuras o sustancias que se deseen investigar en la muestra correspondiente. Figura 1-1. Un órgano es transformado en una película transparente. - Pág. 5 - CAP 1: TÉCNICAS MICROSCÓPICAS Los pasos de las técnicas microscópicas para obtener un preparado histológico permanente (láminas) son: 1. Toma de la muestra. 2. Fijación. 3. Inclusión. 4. Microtomía. 5. Coloración. 1. TOMA DE LA MUESTRA Es el momento que se selecciona el órgano o tejido a estudiar. De tres fuentes puede provenir el material humano: las necropsias, las biopsias y las piezas operadas. De éstas, sólo la primera puede darnos material normal; las dos últimas habitualmente proporcionarán tejidos para estudio histopatológico. - Necropsias: son las piezas que se obtienen de un cadáver. Para histología normal es necesario que se trate de un cadáver fresco y que no haya sido atacado por ninguna lesión, por lo menos el órgano que se quiere estudiar. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Diversity of Adult Neural Stem and Progenitor Cells in Physiology and Disease
cells Review Diversity of Adult Neural Stem and Progenitor Cells in Physiology and Disease Zachary Finkel, Fatima Esteban, Brianna Rodriguez, Tianyue Fu, Xin Ai and Li Cai * Department of Biomedical Engineering, Rutgers University, Piscataway, NJ 08854, USA; [email protected] (Z.F.); [email protected] (F.E.); [email protected] (B.R.); [email protected] (T.F.); [email protected] (X.A.) * Correspondence: [email protected] Abstract: Adult neural stem and progenitor cells (NSPCs) contribute to learning, memory, main- tenance of homeostasis, energy metabolism and many other essential processes. They are highly heterogeneous populations that require input from a regionally distinct microenvironment including a mix of neurons, oligodendrocytes, astrocytes, ependymal cells, NG2+ glia, vasculature, cere- brospinal fluid (CSF), and others. The diversity of NSPCs is present in all three major parts of the CNS, i.e., the brain, spinal cord, and retina. Intrinsic and extrinsic signals, e.g., neurotrophic and growth factors, master transcription factors, and mechanical properties of the extracellular matrix (ECM), collectively regulate activities and characteristics of NSPCs: quiescence/survival, prolifer- ation, migration, differentiation, and integration. This review discusses the heterogeneous NSPC populations in the normal physiology and highlights their potentials and roles in injured/diseased states for regenerative medicine. Citation: Finkel, Z.; Esteban, F.; Keywords: central nervous system (CNS); ependymal cells; neural stem and progenitor cells (NSPC); Rodriguez, B.; Fu, T.; Ai, X.; Cai, L. NG2+ cells; neurodegenerative diseases; regenerative medicine; retina injury; spinal cord injury Diversity of Adult Neural Stem and (SCI); traumatic brain injury (TBI) Progenitor Cells in Physiology and Disease. Cells 2021, 10, 2045. -

11 Introduction to the Nervous System and Nervous Tissue
11 Introduction to the Nervous System and Nervous Tissue ou can’t turn on the television or radio, much less go online, without seeing some- 11.1 Overview of the Nervous thing to remind you of the nervous system. From advertisements for medications System 381 Yto treat depression and other psychiatric conditions to stories about celebrities and 11.2 Nervous Tissue 384 their battles with illegal drugs, information about the nervous system is everywhere in 11.3 Electrophysiology our popular culture. And there is good reason for this—the nervous system controls our of Neurons 393 perception and experience of the world. In addition, it directs voluntary movement, and 11.4 Neuronal Synapses 406 is the seat of our consciousness, personality, and learning and memory. Along with the 11.5 Neurotransmitters 413 endocrine system, the nervous system regulates many aspects of homeostasis, including 11.6 Functional Groups respiratory rate, blood pressure, body temperature, the sleep/wake cycle, and blood pH. of Neurons 417 In this chapter we introduce the multitasking nervous system and its basic functions and divisions. We then examine the structure and physiology of the main tissue of the nervous system: nervous tissue. As you read, notice that many of the same principles you discovered in the muscle tissue chapter (see Chapter 10) apply here as well. MODULE 11.1 Overview of the Nervous System Learning Outcomes 1. Describe the major functions of the nervous system. 2. Describe the structures and basic functions of each organ of the central and peripheral nervous systems. 3. Explain the major differences between the two functional divisions of the peripheral nervous system.