Highly Polar Bonds and the Meaning of Covalency and Ionicity—Structure and Bonding of Alkali Metal Hydride Oligomers F
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -

United States Patent Office Patented Aug
3,459,514 United States Patent Office Patented Aug. 5, 1969 2 quired is smaller than the requirements of the prior art. 3,459,514 It is still a further object of the present invention to pro METHOD FOR PREPARING ALKAL vide a process whereby the amount of by-product pro METAL BOROHYDRDES James D. Johnston and Albert P. Giraitis, Baton Rouge, duced is less than that produced by the prior art. Other La., assignors to Ethyl Corporation, New York, N.Y., a objects will become apparent from the ensuing descrip corporation of Virginia tion. No Drawing. Continuation-in-part of applications Ser. No. The above objects are accomplished by the provision 308,691, Sept. 13, 1963, and Ser. No. 322,054, Nov. 7, of a process for producing an alkali metal borohydride 1963. This application Oct. 1, 1964, Ser. No. 400,888 which comprises reacting together an alkali metal hy ret, C. C01b 6/14 O dride, desiccated borax, hydrogen, and silicon at a tem U.S. C. 23-362 6 Claims perature within the range of from about 200 C. to about 900° C. The alkali metal present during the course of the re ABSTRACT OF THE DISCLOSURE action is a member selected from Group I-A of the A method of preparing alkali metal borohydrides com Periodic Chart of the Elements, Fisher Scientific Com prising reacting an alkali metal or alkali metal hydride, pany, 1955. The alkali metals include lithium, sodium, desiccated borax, hydrogen, and silicon, in an inert hy potassium, rubidium, and cesium. Sodium is a preferred drocarbon at about 250° C. -

Sop Pyrophoric 2 12/16/2019
Owner DOC. NO. REV. DATE C.H.O SOP PYROPHORIC 2 12/16/2019 DOC. TITLE SOP FOR PYROPHORIC CHEMICALS Environmental Health & Safety STANDARD OPERATING PROCEDURES (SOP) FOR WORKING WITH PYROPHORIC CHEMICALS AT AMHERST COLLEGE ___________________________________________________________________ General Information Pyrophoric Chemicals are solid, liquid, or gas compounds that, when exposed to air or moisture at or below 54°C (130°F), can spontaneously ignite. Examples of Pyrophoric chemicals used at Amherst College Laboratories include: sodium hydride, zinc powder, and Grignard reagents. See the “Appendix” page below for a full list of Pyrophoric Chemicals. Pyrophoric chemicals are often used as catalysts in chemical reactions or as reducing and deprotonating agents in organic chemistry. Note that Pyrophoric chemicals may also be characterized by other hazards, hence, users of these chemicals may also need to refer to other SOPs that cover other hazards. In addition, each individual chemical’s Safety Data Sheet (SDS) should be consulted before they are used. _____________________________________________________________________________________ Personal Protective Equipment When working with Pyrophoric Chemicals, the following personal protective equipment (PPE) must be worn, at a minimum. Depending on the specific chemical, other forms of protection might be required. Consult the SDS for each chemical before use: Splash goggles Lab coat (Fire resistant lab coat highly recommended) Long pants Close toed shoes Gloves – Nitrile gloves adequate for accidental contact with small quantities. However, the use of fire resistant Nomex/ Leather Pilot’s gloves is highly recommended _____________________________________________________________________________________ Safety Devices All work with Pyrophoric chemicals must be done in a glove box, vacuum manifold, or any enclosed inert environment. If work must be done in a fume hood, ensure that the hood sash is in the lowest feasible position. -

Innovations Enabled by the U.S. Department of Energy Fuel Cell Technologies Office
Pathways to Success: Innovations Enabled by the U.S. Department of Energy Fuel Cell Technologies Office November 2018 Prepared by Pacific Northwest National Laboratory for the U.S. Department of Energy Fuel Cell Technologies Office i Notice This report is being disseminated by the U.S. Department of Energy (DOE). As such, this document was prepared in compliance with Section 515 of the Treasury and General Government Appropriations Act for Fiscal Year 2001(Public Law 106-554) and information quality guidelines issued by DOE. Though this report does not constitute “influential” information, as that term is defined in DOE’s information quality guidelines or the Office of Management and Budget’s Information Quality Bulletin for Peer Review, the report was reviewed both internally and externally prior to publication. Reviewers included technical experts from Pacific Northwest National Laboratory and DOE’s Fuel Cell Technologies Office. The views and opinions of the authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. ii Table of Contents Table of Contents ...................................................................................................... -

Back Matter (PDF)
INDEX TO VOLUME 171 (A) Adiabatic piezo-optic coefficient of liquids (Raman and Venkataraman), 137. Allen, J. F. and Ganz, E. The influence of pressure on the thermal conductivity of liquid He n, 242. Appleton, E. V. and Weekes, K. On lunar tides in the upper atmosphere, 171. Arnot, F. L. and Hart, W. D. Electron transfer in argon, 383. Arnot, F. L. and M’Ewen, M. B. The formation of helium molecules, II, 106. Braddick, H. J. J. Cosmic ray bursts at high altitude, 314. Bruce, H. D. Optical thickness of the transition layer between transparent media, 411. Brookman, E. F. See Norrish and Brookman. Budden, K. G., Ratcliffe, J. A. and Wilkes, M. V. Further investigations of very long waves reflected from the ionosphere, 188. Burgoyne, J. H. The combustion of aromatic and alicyclic hydrocarbons, II, 421. Carmichael, H. and Dymond, E. G. High altitude cosmic radiation measure ments near the north geomagnetic pole, 321. ------------- Upper air investigations in North-West Greenland, 346. Combustion of aromatic and alicyclic hydrocarbons (Burgoyne), 421. Conn, G. K. T. and Twigg, G. H. Infra-red analysis applied to the exchange reaction between ethylene and deuteroethylene, 70. Copper-cuprous oxide photocells (Mott), 281. Coriolis perturbations in methane spectrum (Jahn), 450. Cosmic radiation near the north pole (Carmichael and Dymond), 321. Cosmic ray bursts at high altitude (Braddick), 314. Coulomb law, deviation for the proton (Frdhlich, Heitler and Kahn), 269. Crystal rectifiers, theory (Mott), 27. Davoud, J. G. and Hinshelwood, C. N. The thermal decomposition of diethyl ether, 39. Diamagnetic anisotropy of organic molecules (Lonsdale), 541. -

Molecular Hydrogen and Catalytic Combustion in the Production Of
Molecular Hydrogen and Catalytic Combustion in the Production of Hyperpolarized 83Kr and 129Xe MRI Contrast Agents. Nicola J. Rogers1,a, Fraser Hill-Casey1,2, Karl F. Stupic1, b, Joseph S. Six1,c, Clementine Lesbats1, Sean P. Rigby2, Jacques Fraissard3, Galina E. Pavlovskaya1, and Thomas Meersmann1*. 1Sir Peter Mansfield Imaging Centre, University of Nottingham, Nottingham, NG7 2RD, United Kingdom 2Department of Chemical and Environmental Engineering, University of Nottingham, Nottingham NG7 2RD, United Kingdom 3Université Pierre et Marie Curie, Ecole Supérieure de Physique et Chimie Industrielles, 75005 Paris, France aCurrent address; Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, United Kingdom bCurrent address: Division of Magnetics, National Institute of Standards and Technology, Boulder, Colorado, USA, 80305. cCurrent address: Carestream Health Inc., 8124 Pacific Avenue, White City, Oregon, 97503, USA *Author to whom correspondence should be addressed. Electronic mail: [email protected] Keywords: spin exchange optical pumping, hyperpolarized noble gas contrast agents, cryogenic separation, chemical looping combustion, catalytic hydrogen oxidation Classification: Physical Sciences; Applied Physical Sciences. Argon40_vsN29b 2016_01_11 1 Abstract. Hyperpolarized (hp) 83Kr is a promising MRI contrast agent for the diagnosis of pulmonary diseases affecting the surface of the respiratory zone. However, the distinct physical properties of 83Kr that enable unique MRI contrast also complicate the production of hp 83Kr. This work presents a radically new approach in the generation of hp 83Kr that can likewise be utilized for the production of hp 129Xe. Molecular nitrogen, typically used as buffer gas in spin exchange optical pumping (SEOP), was replaced by molecular hydrogen without penalty for the achievable hyperpolarization. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Catalytic Tuning of Sorption Kinetics of Lightweight Hydrides: a Review of the Materials and Mechanism
catalysts Review Catalytic Tuning of Sorption Kinetics of Lightweight Hydrides: A Review of the Materials and Mechanism Ankur Jain 1,*, Shivani Agarwal 2 and Takayuki Ichikawa 3,* 1 Natural Science Centre for Basic Research & Development, Hiroshima University, Higashihiroshima 739-8530, Japan 2 Department of Physics, JECRC University, Jaipur 303905, India; [email protected] 3 Graduate School of Engineering, Hiroshima University, Higashihiroshima 739-8527, Japan * Correspondence: [email protected] or [email protected] (A.J.); [email protected] (T.I.); Tel.: +81-82-424-4609 (A.J.); +81-82-424-5744 (T.I.) Received: 31 October 2018; Accepted: 5 December 2018; Published: 11 December 2018 Abstract: Hydrogen storage materials have been a subject of intensive research during the last 4 decades. Several developments have been achieved in regard of finding suitable materials as per the US-DOE targets. While the lightweight metal hydrides and complex hydrides meet the targeted hydrogen capacity, these possess difficulties of hard thermodynamics and sluggish kinetics of hydrogen sorption. A number of methods have been explored to tune the thermodynamic and kinetic properties of these materials. The thermodynamic constraints could be resolved using an intermediate step of alloying or by making reactive composites with other hydrogen storage materials, whereas the sluggish kinetics could be improved using several approaches such as downsizing and the use of catalysts. The catalyst addition reduces the activation barrier and enhances the sorption rate of hydrogen absorption/desorption. In this review, the catalytic modifications of lightweight hydrogen storage materials are reported and the mechanism towards the improvement is discussed. -

Nomenclature of Inorganic Compounds
WORKSHEET GENERATORS www.vaxasoftware.com/pc Exercises of Nomenclature of inorganic compounds 1) Give the formula for each of the following: a) beryllium monoxide, beryllium oxide b) water c) dialuminium trioxide, aluminium oxide d) manganese monoxide, manganese(II) oxide e) disodium dioxide, sodium peroxide f) carbon monoxide, carbon(II) oxide 2) Name each of the following compounds: a) Ag2O2 b) CoO c) CdO d) BaO2 e) H2O f) CaO 3) Give the formula for each of the following: a) calcium hydride b) zinc hydride c) aluminium hydride d) phosphine e) francium hydride f) sodium hydride 4) Name each of the following compounds: a) AgH b) CsH c) AsH3 d) BaH2 e) RbH f) KH 5) Give the formula for each of the following: a) nickel(II) hydroxide b) magnesium hydroxide c) copper(I) hydroxide d) caesium hydroxide e) rubidium hydroxide f) calcium hydroxide 6) Name each of the following compounds: a) NaOH b) NH4OH c) Cr(OH)3 d) Fe(OH)2 e) Cr(OH)2 f) Cu(OH)2 1 WORKSHEET GENERATORS www.vaxasoftware.com/pc Exercises of Nomenclature of inorganic compounds 7) Give the formula for each of the following: a) rubidium bromide b) copper(I) chloride c) hydrogen iodide d) beryllium bromide e) dinitrogen triselenide, nitrogen(III) selenide, nitrous selenide f) hydrogen chloride, hydrochloric acid 8) Name each of the following compounds: a) HF b) H2Se(ac) c) NH4Cl d) LiCl e) As2S3 f) HI(ac) 9) Give the formula for each of the following: a) hydrogen chromate, chromic acid b) hydrogen orthophosphate, orthophosphoric acid c) hydrogen seleniate, selenic acid d) hydrogen -

US5354918.Pdf
|||||||||||||||||US005354918A United States Patent 19 11) Patent Number: 5,354,918 Ohsaki et al. 45) Date of Patent: Oct. 11, 1994 (54 HIGHLY PURE MONOALKYLPHOSPHINE OTHER PUBLICATIONS (75) Inventors: Hiromi Ohsaki; Kazuyuki Asakura, Hoff et al., Journal of Organic Chemistry, (24) pp. both of Joetsu: Masashi Sugiya, 356-359, 1959. Tokyo; Yutaka Demura; Tomio Chemical Abstracts, vol. 117, 1992, Columbus, Ohio, Yanai, both of Kohriyama, all of US; abstract No. 171693s, & JP-A-04 149 187 (Nippon Japan Mining Co., Ltd.). Primary Examiner-Johann Richter 73) Assignees: Shin-Etsu Chemical Co., Ltd.; Nippon Assistant Examiner-Margaret J. Page Chemical Industrial Co., Ltd., both of Japan Attorney, Agent, or Firm-Parkhurst, Wendel & Rossi 57 ABSTRACT (21) Appl. No.: 91,363 The method for preparing a monoalkylphosphine of high purity comprises reacting phosphine and an alkene (22 Filed: Jul. 15, 1993 in the presence of an anhydrous alkanesulfonic acid as a catalyst in a solvent having a boiling point higher than that of the monoalkylphosphine produced. The result 30 Foreign Application Priority Data ing monoalkylphosphine is brought into contact with an Jul. 17, 1992 JP Japan .................................. 4-1904.87 alkali solution to remove the catalyst, which is a sulfur Nov. 9, 1992 JP Japan .... ... 4-298629 atom-containing compound, remaining in the solution. Mar. 12, 1993 JP Japan .................................... 5-51806 The reaction solution from which the remaining cata Mar. 22, 1993 JP Japan .................................... 5-61775 lyst is removed is then brought into contact with alkali metal hydrides or alkaline earth metal hydrides to elimi 51) Int. Cl. ................................................ C07F 9/50 nate any moisture remaining in the reaction solution. -

Acetylene (Dissolved)
ACETYLENE (DISSOLVED) A-Gas South Africa Chemwatch Hazard Alert Code: 4 Chemwatch: 1001 Issue Date: 16/10/2014 Version No: 5.1.1.1 Print Date: 16/08/2018 Safety Data Sheet (Conforms to Regulaon (EU) No 2015/830) S.REACH.GBR.EN SECTION 1 IDENTIFICATION OF THE SUBSTANCE / MIXTURE AND OF THE COMPANY / UNDERTAKING 1.1. Product Idenfier Product name ACETYLENE (DISSOLVED) Chemical Name acetylene Synonyms C2H2; acetylene dissolved; ethine; ethyne; acetylen Proper shipping name ACETYLENE, DISSOLVED Chemical formula C2H2 Other means of idenficaon Not Available CAS number 74-86-2 EC number 200-816-9 Index number 601-015-00-0 REACH registraon number 01-2119457406-36-XXXX 1.2. Relevant idenfied uses of the substance or mixture and uses advised against Fuel gas for oxy-acetylene welding, flame cung, gouging, metal scale removal, brazing, flame hardening and general localised heang. Burnt Relevant idenfied uses with oxygen as illuminant in navigaonal beacons. Used as a source of carbon black and as reactant in manufacture of vinyl chloride, acrylates etc. Uses advised against Not Applicable 1.3. Details of the supplier of the safety data sheet Registered company name A-Gas South Africa Address 8 Railway Road Montague Gardens 7441 Cape Town South Africa Telephone +27 0 21 551 8790 Fax +27 0 21 551 8758 Website Not Available Email Not Available 1.4. Emergency telephone number Associaon / Organisaon Not Available Emergency telephone Not Available numbers Other emergency telephone Not Available numbers SECTION 2 HAZARDS IDENTIFICATION 2.1. Classificaon of the substance or mixture Classificaon according to regulaon (EC) No 1272/2008 H220 - Flammable Gas Category 1, H280 - Gas under Pressure [CLP] [2] Legend: 1. -
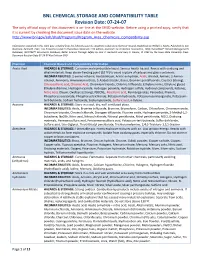
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers