Scientific Advisory Board
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Theoretical Study of Sarin Adsorption On
Chemical Physics Letters 738 (2020) 136816 Contents lists available at ScienceDirect Chemical Physics Letters journal homepage: www.elsevier.com/locate/cplett Research paper Theoretical study of sarin adsorption on (12,0) boron nitride nanotube doped with silicon atoms T ⁎ ⁎ Jeziel Rodrigues dos Santosa, , Elson Longo da Silvab, Osmair Vital de Oliveirac, , José Divino dos Santosa a Universidade Estadual de Goiás, Campus Anápolis, CEP: 75.132-903 GO, Brazil b INCTMN, LIEC, Departamento de Química da Universidade Federal de São Carlos, CEP: 13.565-905 São Carlos, SP, Brazil c Instituto Federal de Educação, Ciência e Tecnologia de São Paulo, Campus Catanduva, CEP: 15.808-305 Catanduva, SP, Brazil HIGHLIGHTS • DFT method was used to study the adsorption of nerve agent sarin by BNNT. • Electronic properties of pristine BNNT are improved by Si impurity atoms. • The adsorption of sarin by Si-doped BNNT is highest favorable than the pure BNNT. • Si-doped BNNT can be a new gas sensor for sarin gas detection and its derivatives. ARTICLE INFO ABSTRACT Keywords: Sarin gas is one of the most lethal nerve agent used in chemical warfare, which its detection is import to prevent Nerve agent sarin a chemical attack and to identify a contamination area. Herein, density functional theory was used to investigate Gas sensor the (12,0) boron nitride nanotube (BNNT) and Si–doped BNNT as possible candidates to sarin detection. The Si- Boron nitride nanotube atoms doped improve the electronic properties of nanotubes by altering the electrostatic potential, HOMO and DFT LUMO energies. Based in the adsorption energies and the conductivity increased to ~33 and 350%, respectively, for Si- and 2Si-BNNT imply that they can be used for sarin detection. -

Decontamination of Agent Yellow, a Lewisite and Sulfur Mustard Mixture
EPA 600/R-14/436 | March 2015 | www.epa.gov/research Decontamination of Agent Yellow, a Lewisite and Sulfur Mustard Mixture Office of Research and Development National Homeland Security Research Center Decontamination of Agent Yellow, a Lewisite and Sulfur Mustard Mixture Evaluation Report National Homeland Security Research Center Office of Research and Development U.S. Environmental Protection Agency Research Triangle Park, NC 27711 ii Disclaimer The United States Environmental Protection Agency through its Office of Research and Development’s National Homeland Security Research Center funded and managed the research described here under EPA Contract Number EP-C-10-001, Work Assignment Number 4-28 with Battelle. This report has been peer and administratively reviewed and has been approved for publication as an Environmental Protection Agency report. It does not necessarily reflect views of the Environmental Protection Agency. No official endorsement should be inferred. The Environmental Protection Agency does not endorse the purchase or sale of any commercial products or services. Questions concerning this document or its application should be addressed to: Lukas Oudejans, Ph.D. Decontamination and Consequence Management Division National Homeland Security Research Center Office of Research and Development U.S. Environmental Protection Agency (MD-E343-06) 109 T.W. Alexander Drive Research Triangle Park, NC 27711 Phone: 919-541-2973 Fax: 919-541-0496 E-mail: [email protected] iii Acknowledgments The following individuals are acknowledged -

Warfare Agents for Modeling Airborne Dispersion in and Around Buildings
LBNL-45475 ERNEST ORLANDO LAWRENCE BERKELEY NATIn NAL LABORATORY Databaseof Physical,Chemicaland ToxicologicalPropertiesof Chemical and Biological(CB)WarfitreAgentsfor ModelingAirborneDispersionIn and AroundBuildings TracyThatcher,RichSextro,andDonErmak Environmental Energy Technologies Division DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor The Regents of the University of Catifomia, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of anY information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommend at i on, or favoring by the United States Government or any agency thereof, or The Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof, or The Regents of the University of California. Ernest Orlando Lawrence Berkeley National Laboratory is an equal opportunity employer. DISCLAIMER Portions of this document may be illegible in electronic image products. Images are produced -

Medicine with Deeper, More Biochemical Conception Of
Edinburgh Medical Journal May 1951 APPLICATIONS of chemical defence research IN MEDICINET? By Professor R. A. PETERS to be invited It is a great responsibility, as well as a great honour, to Cameron lecturers give this lecture ; and I expect that with other I this ancient seat of share a sense of awe upon standing here in is learning. For me it is an awe tinged with much feeling (it possible that this is partly because some forebears of my mother came from near to recent of the this ; it is in part due the perusal city) perhaps " address here Froude in on the Times of John Knox, given by 1865 " " entitled Influence of the Reformation upon the Scottish Character ; this address drives home to the Sassenach the sterner stuff of which all of us in the the Scots are made ; but I really think it is more that as south, even if we come as I do from a Medical School as ancient the Hospital of St Bartholomew, know full well that Edinburgh stands high indeed in the world of Medical Schools as Scotland itself stands in the world of intellect. Elsewhere I have discussed fully the historical background of our for me back researches upon Chemical Defence substances, which go to World War I.23 Again in my Dixon lecture,24 I have indicated some of the interest for pharmacology. The outlook for medicine has not the been reviewed so far. Therefore I propose to-day to discuss of British influence upon the future of therapeutics of the discovery anti-lewisite and of some of the other researches upon war gas but substances ; and to consider not only the leads towards therapy, the possible newer trends given to medicine itself. -

Information Data Sheet Detector Tube QUALITEST QL Part No. (US): 497665 Part No. D5085810
Information Data Sheet Detector Tube QUALITEST QL Part No. (US): 497665 Part No. D5085810 1. Application Detction of dangerous (toxic or combustible) gases and vapors in air.In particular fot testing the air in confined spaces such as fuel tanks, storage bins, cable vaults, sewers. Furthermore the tube QL can be used to detect or localize leaks, e.g. in pipelines. 2. General Description The detector tube QL does not have any calibration scale. The indication is non-specific and qualita- tive, i. e. only the presence resp. the absence of contaminants will be detected. The indication gives no information about type or concentration of contaminants detected. A special detector tube with quantitative indication shoud be used if the presence or the concentration of a known contaminant is to be determined. This applies also to substances which can not be indicated by the detector tube QL. The various contaminants are indicated with different sensitivity. However, the detector tube is sufficiently sensitive for all listed substances. Normally, concentrations of a few ppm are detectable. Among others, the following substances are indicated: Acetone, acetylene, benzene, 1.3-butadiene, butanes, butylenes, carbon disulfide, carbon monoxide, cyclohexane, diesel oil, ethanol (ethyl alcohol), ethylene, formic acid, fuel oil, gasoline (engine fuel),hydrogen chloride, hydrogen sulfide, kerosene, liquid petroleum gas (propane, butanes), methyl ethyl ketone (butanone), pentanes and other saturated hydrocarbons, phenol, propane, propanols (propyl alcohols), -
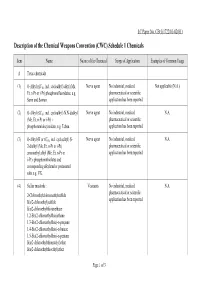
Description of the Chemical Weapons Convention (CWC) Schedule 1 Chemicals
LC Paper No. CB(1)1722/01-02(01) Description of the Chemical Weapons Convention (CWC) Schedule 1 Chemicals Item Name Nature of the Chemical Scope of Application Examples of Common Usage A Toxic chemicals (1) O-Alkyl (≤C10, incl. cycloalkyl) alkyl (Me, Nerve agent No industrial, medical, Not applicable (N.A.) Et, n-Pr or i-Pr) phosphonofluoridates, e.g. pharmaceutical or scientific Sarin and Soman. application has been reported. (2) O-Alkyl (≤C10, incl. cycloalkyl) N,N-dialkyl Nerve agent No industrial, medical, N.A. (Me, Et, n-Pr or i-Pr) - pharmaceutical or scientific phosphoramidocyanidate, e.g. Tabun. application has been reported. (3) O-Alkyl (H or ≤C10, incl. cycloalkyl) S- Nerve agent No industrial, medical, N.A. 2-dialkyl (Me, Et, n-Pr or i-Pr) pharmaceutical or scientific aminoethyl alkyl (Me, Et, n-Pr or application has been reported. i-Pr)- phosphonothiolates and corresponding alkylated or protonated salts e.g. VX. (4) Sulfur mustards : Vesicants No industrial, medical, N.A. pharmaceutical or scientific 2-Chloroethylchloromethylsulfide application has been reported. Bis(2-chloroethyl)sulfide Bis(2-chloroethylthio)methane 1,2-Bis(2-chloroethylthio)ethane 1,3-Bis(2-chloroethylthio)-n-propane 1,4-Bis(2-chloroethylthio)-n-butane 1,5-Bis(2-chloroethylthio)-n-pentane Bis(2-chloroethylthiomethyl)ether Bis(2-chloroethylthioethyl)ether Page 1 of 3 Item Name Nature of the Chemical Scope of Application Examples of Common Usage (5) Lewisites : Vesicants No industrial, medical, N.A. pharmaceutical or scientific Lewisite 1 : 2-Chlorovinyldichloroarsine application has been reported. Lewisite 2 : Bis(2-chlorovinyl)chloroarsine Lewisite 3 : Tris(2-chlorovinyl)arsine (6) Nitrogen mustards : Vesicants The chemical has medical Only HN2 has been reported to application. -

"The Science for Diplomats" Annex on Chemicals
ORGANISATION FOR THE PROHIBITION OF CHEMICAL WEAPONS "THE SCIENCE FOR DIPLOMATS" ANNEX ON CHEMICALS A user friendly and scientifically annotated version of the Chemical Weapons Convention Annex on Chemicals OPCW THE “SCIENCE FOR DIPLOMATS” ANNEX ON CHEMICALS A user friendly and scientifically annotated version of the Chemical Weapons Convention Annex on Chemicals1 CONTENTS A. GUIDELINES FOR SCHEDULES OF CHEMICALS B. VISUALISING AND READING MOLECULAR STRUCTURES C. SCHEDULES OF CHEMICALS D. RIOT CONTROL AGENTS 1 An official version of the Annex on Chemicals can be obtained from the OPCW public website, www.opcw.org/chemical-weapons-convention/annexes/annex-chemicals/annex-chemicals. Version 3.0 – 10 March 2019 A. GUIDELINES FOR SCHEDULES OF CHEMICALS Guidelines for Schedule 1 1. The following criteria shall be taken into account in considering whether a toxic chemical or precursor should be included in Schedule 1: (a) It has been developed, produced, stockpiled or used as a chemical weapon as defined in Article II; (b) It poses otherwise a high risk to the object and purpose of this Convention by virtue of its high potential for use in activities prohibited under this Convention because one or more of the following conditions are met: (i) It possesses a chemical structure closely related to that of other toxic chemicals listed in Schedule 1, and has, or can be expected to have, comparable properties; (ii) It possesses such lethal or incapacitating toxicity as well as other properties that would enable it to be used as a chemical weapon; (iii) It may be used as a precursor in the final single technological stage of production of a toxic chemical listed in Schedule 1, regardless of whether this stage takes place in facilities, in munitions or elsewhere; (c) It has little or no use for purposes not prohibited under this Convention. -

Nerve Agent - Lntellipedia Page 1 Of9 Doc ID : 6637155 (U) Nerve Agent
This document is made available through the declassification efforts and research of John Greenewald, Jr., creator of: The Black Vault The Black Vault is the largest online Freedom of Information Act (FOIA) document clearinghouse in the world. The research efforts here are responsible for the declassification of MILLIONS of pages released by the U.S. Government & Military. Discover the Truth at: http://www.theblackvault.com Nerve Agent - lntellipedia Page 1 of9 Doc ID : 6637155 (U) Nerve Agent UNCLASSIFIED From lntellipedia Nerve Agents (also known as nerve gases, though these chemicals are liquid at room temperature) are a class of phosphorus-containing organic chemicals (organophosphates) that disrupt the mechanism by which nerves transfer messages to organs. The disruption is caused by blocking acetylcholinesterase, an enzyme that normally relaxes the activity of acetylcholine, a neurotransmitter. ...--------- --- -·---- - --- -·-- --- --- Contents • 1 Overview • 2 Biological Effects • 2.1 Mechanism of Action • 2.2 Antidotes • 3 Classes • 3.1 G-Series • 3.2 V-Series • 3.3 Novichok Agents • 3.4 Insecticides • 4 History • 4.1 The Discovery ofNerve Agents • 4.2 The Nazi Mass Production ofTabun • 4.3 Nerve Agents in Nazi Germany • 4.4 The Secret Gets Out • 4.5 Since World War II • 4.6 Ocean Disposal of Chemical Weapons • 5 Popular Culture • 6 References and External Links --------------- ----·-- - Overview As chemical weapons, they are classified as weapons of mass destruction by the United Nations according to UN Resolution 687, and their production and stockpiling was outlawed by the Chemical Weapons Convention of 1993; the Chemical Weapons Convention officially took effect on April 291997. Poisoning by a nerve agent leads to contraction of pupils, profuse salivation, convulsions, involuntary urination and defecation, and eventual death by asphyxiation as control is lost over respiratory muscles. -

Lewisite Fact Sheet
Lewisite Fact Sheet HIGHLIGHTS: It is unlikely that the general population will be exposed to blister agents Lewisite or Mustard-Lewisite. People who breathe in vapors of Lewisite or Mustard-Lewisite may experience damage to the respiratory system. Contact with the skin or eye can result in serious burns. Lewisite or Mustard-Lewisite also can cause damage to bone marrow and blood vessels. Exposure to high levels may be fatal. Blister agents Lewisite and Mustard-Lewisite have not been found in any of the 1,585 National Priorities List sites identified by the Environmental Protection Agency (EPA). What are lewisite and mustard-lewisite? Lewisite is an oily, colorless liquid with an odor like geraniums. Mustard-Lewisite Mixture is a liquid with a garlic-like odor. Mustard-Lewisite is a mixture of Lewisite and a sulfur mustard known as HD. Lewisite might have been used as a chemical weapon by Japan against Chinese forces in the 1930s, but such reports have not been confirmed. Any stored Lewisite in the United States must be destroyed before April 2007, as mandated by the Chemical Weapons Convention. What happens to lewisite and mustard-lewisite when it enters the environment? • Blister agents Lewisite and Mustard-Lewisite could enter the environment from an accidental release. • In air, blister agents Lewisite and Mustard-Lewisite will be broken down by compounds that are found in the air, but they may persist in air for a few days before being broken down. • Lewisite and Mustard-Lewisite will be broken down in water quickly, but small amounts may evaporate. • Lewisite and Mustard-Lewisite will be broken down in moist soil quickly, but small amounts may evaporate. -

Decision-Making in Chemical Warfare Agent (CWA) Response There Is a Lot of Fear Associated with Chemical Will Act Accordingly
Application Note: 102 Decision-Making in Chemical Warfare Agent (CWA) Response There is a lot of fear associated with Chemical will act accordingly. If the first responders Warfare Agents (CWAs). The misnomer over-react and immediately jump into full “Nerve Gas” quickly brings horrible images to encapsulation protection it could panic the the minds of many civilians. But if we lay aside public and cause unnecessary worry and the politics and fear, CWA detection should even injury. treated like other gas/vapor detection challenges. It should be a collaborative Over Protection Can Be Dangerous process encompassing physical clues, threat to the Responder scenario, biological clues, and a variety of Heat stress is the number one injury in sensing technologies. No one clue or HazMat response and immediately jumping technology is always correct. Experience and into full Level A encapsulation is a good way the use of multiple clues and technologies are of overheating oneself. Level A the keys to successful CWA response. encapsulation also makes one much more Understanding what the clues are and how to susceptible to slip, trip and fall injuries. layer them to make a decision is critical to Finally, over protection makes it harder to get successful CWA response. things done. When properly used, detection allows responders to respond at lower levels Why is Gas Detection Important? of Personal Protective Equipment (PPE) to Responders cannot rely on their senses for provide the highest levels of safety to decision-making. Without effectively knowing themselves and to the community that they how to use detection techniques responders protect. -

Warning: the Following Lecture Contains Graphic Images
What the новичок (Novichok)? Why Chemical Warfare Agents Are More Relevant Than Ever Matt Sztajnkrycer, MD PHD Professor of Emergency Medicine, Mayo Clinic Medical Toxicologist, Minnesota Poison Control System Medical Director, RFD Chemical Assessment Team @NoobieMatt #ITLS2018 Disclosures In accordance with the Accreditation Council for Continuing Medical Education (ACCME) Standards, the American Nurses Credentialing Center’s Commission (ANCC) and the Commission on Accreditation for Pre-Hospital Continuing Education (CAPCE), states presenters must disclose the existence of significant financial interests in or relationships with manufacturers or commercial products that may have a direct interest in the subject matter of the presentation, and relationships with the commercial supporter of this CME activity. The presenter does not consider that it will influence their presentation. Dr. Sztajnkrycer does not have a significant financial relationship to report. Dr. Sztajnkrycer is on the Editorial Board of International Trauma Life Support. Specific CW Agents Classes of Chemical Agents: The Big 5 The “A” List Pulmonary Agents Phosgene Oxime, Chlorine Vesicants Mustard, Phosgene Blood Agents CN Nerve Agents G, V, Novel, T Incapacitating Agents Thinking Outside the Box - An Abbreviated List Ammonia Fluorine Chlorine Acrylonitrile Hydrogen Sulfide Phosphine Methyl Isocyanate Dibotane Hydrogen Selenide Allyl Alcohol Sulfur Dioxide TDI Acrolein Nitric Acid Arsine Hydrazine Compound 1080/1081 Nitrogen Dioxide Tetramine (TETS) Ethylene Oxide Chlorine Leaks Phosphine Chlorine Common Toxic Industrial Chemical (“TIC”). Why use it in war/terror? Chlorine Density of 3.21 g/L. Heavier than air (1.28 g/L) sinks. Concentrates in low-lying areas. Like basements and underground bunkers. Reacts with water: Hypochlorous acid (HClO) Hydrochloric acid (HCl). -

Kinetics of Coal Fly Ash Chlorination by Phosgene Douglas John Adelman Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1984 Kinetics of coal fly ash chlorination by phosgene Douglas John Adelman Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Chemical Engineering Commons Recommended Citation Adelman, Douglas John, "Kinetics of coal fly ash chlorination by phosgene " (1984). Retrospective Theses and Dissertations. 8971. https://lib.dr.iastate.edu/rtd/8971 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This reproduction was made from a copy of a document sent to us for microfilming. While the most advanced technology has been used to photograph and reproduce this document, the quality of the reproduction is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help clarify markings or notations which may appear on this reproduction. 1.The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting through an image and duplicating adjacent pages to assure complete continuity. 2. When an image on the film is obliterated with a round black mark, it is an indication of either blurred copy because of movement during exposure, duplicate copy, or copyrighted materials that should not have been filmed.