Regulation of Cell Polarity and Invasion by TGF-Β and BMP Signaling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amh) Relative to Sox9a, Sox9b, and Cyp19a1a, During Gonad Development
Gene Expression Patterns 5 (2005) 655–667 www.elsevier.com/locate/modgep Characterization and expression pattern of zebrafish anti-Mu¨llerian hormone (amh) relative to sox9a, sox9b, and cyp19a1a, during gonad development Adriana Rodrı´guez-Marı´, Yi-Lin Yan, Ruth A. BreMiller, Catherine Wilson, Cristian Can˜estro, John H. Postlethwait* Institute of Neuroscience, University of Oregon, 1425 E. 13th Avenue, Eugene, OR 97403, USA Received 28 January 2005; received in revised form 28 February 2005; accepted 28 February 2005 Available online 19 April 2005 Abstract The role of Anti-Mu¨llerian hormone (Amh) during gonad development has been studied extensively in mammals, but is less well understood in other vertebrates. In male mammalian embryos, Sox9 activates expression of Amh, which initiates the regression of the Mu¨llerian ducts and inhibits the expression of aromatase (Cyp19a1), the enzyme that converts androgens to estrogens. To better understand shared features of vertebrate gonadogenesis, we cloned amh cDNA from zebrafish, characterized its genomic structure, mapped it, analyzed conserved syntenies, studied its expression pattern in embryos, larvae, juveniles, and adults, and compared it to the expression patterns of sox9a, sox9b and cyp19a1a. We found that the onset of amh expression occurred while gonads were still undifferentiated and sox9a and cyp19a1a were already expressed. In differentiated gonads of juveniles, amh showed a sexually dimorphic expression pattern. In 31 days post-fertilization juveniles, testes expressed amh and sox9a, but not cyp19a1a, while ovaries expressed cyp19a1a and sox9b, but not amh.In adult testes, amh and sox9a were expressed in presumptive Sertoli cells. In adult ovaries, amh and cyp19a1a were expressed in granulosa cells surrounding the oocytes, and sox9b was expressed in a complementary fashion in the ooplasm of oocytes. -

The Nuclear Receptor REVERB Represses the Transcription of Growthdifferentiation Factor 10 and 15 Genes in Rat Endometrium Strom
Physiological Reports ISSN 2051-817X ORIGINAL RESEARCH The nuclear receptor REV-ERBa represses the transcription of growth/differentiation factor 10 and 15 genes in rat endometrium stromal cells Lijia Zhao1, Keishiro Isayama1, Huatao Chen1,*, Nobuhiko Yamauchi1, Yasufumi Shigeyoshi2, Seiichi Hashimoto3 & Masa-aki Hattori1 1 Department of Animal and Marine Bioresource Sciences, Graduate School of Agriculture, Kyushu University, Fukuoka, Japan 2 Department of Anatomy and Neurobiology, Kinki University School of Medicine, Osaka, Japan 3 Graduate School of Medicine, The University of Tokyo, Tokyo, Japan Keywords Abstract Circadian clock, decidualization, growth/ differentiation factors, REV-ERBa. Cellular oscillators in the uterus play critical roles in the gestation processes of mammals through entraining of the clock proteins to numerous downstream Correspondence genes, including growth/differentiation factor (Gdf)10 and Gdf15. The expres- Masa-aki Hattori, Department of Animal and sion of Gdf10 and Gdf15 is significantly increased in the uterus during decidu- Marine Bioresource Sciences, Graduate alization, but the mechanism underlying the regulation of Gdf gene expression School of Agriculture, Kyushu University, in the uterus is poorly understood. Here, we focused on the function of the Hakozaki, Higashi-ku, Fukuoka 812-8581, cellular oscillators in the expression of Gdf family by using uterine endome- Japan. Tel: +81-92-642-2938 trial stromal cells (UESCs) isolated from pregnant Per2-dLuc transgenic rats. Fax: +81-92-642-2938 A significant decline of Per2-dLuc bioluminescence activity was induced in E-mail: [email protected] in vitro decidualized UESCs, and concomitantly the expression of canonical clock genes was downregulated. Conversely, the expression of Gdf10 and ⁄ Present address Gdf15 of the Gdf was upregulated. -

A Deletion in GDF7 Is Associated with a Heritable Forebrain Commissural
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.12.091686; this version posted May 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A deletion in GDF7 is associated with a heritable forebrain commissural malformation concurrent with ventriculomegaly and interhemispheric cysts in cats. Yoshihiko Yu1,2,*, Erica K. Creighton1,*, Reuben M. Buckley1, Leslie A. Lyons1,†, 99 Lives Consortium 1Department of Veterinary Medicine and Surgery, College of Veterinary Medicine, University of Missouri, Columbia, MO, 65211, USA 2Laboratory of Veterinary Radiology, Nippon Veterinary and Life Science University, Musashino, Tokyo, 180-8602, Japan *The first two authors (Y.Y., E.K.C) contributed equally to this work. †Corresponding author: Leslie A. Lyons, PhD Department of Veterinary Medicine and Surgery College of Veterinary Medicine University of Missouri Columbia, MO 65211 USA Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.12.091686; this version posted May 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Key words: feline; Felis catus; brain malformation; BMP12; neurodevelopment; genetics; genomics; Mendelian traits; genome-wide association study; whole genome sequencing Abstract An inherited neurologic syndrome in a family of mixed-breed Oriental cats has been characterized as forebrain commissural malformation concurrent with ventriculomegaly and interhemispheric cysts. -

Expression of Mirnas from the Imprinted DLK1/DIO3 Locus Signals the Osteogenic Potential of Human Pluripotent Stem Cells
cells Article Expression of miRNAs from the Imprinted DLK1/DIO3 Locus Signals the Osteogenic Potential of Human Pluripotent Stem Cells Laetitia Barrault 1, Jacqueline Gide 1, Tingting Qing 1, Lea Lesueur 2, Jorg Tost 3 , Jerome Alexandre Denis 2 , Michel Cailleret 2 , Laetitia Aubry 2, Marc Peschanski 1,2, Cécile Martinat 2,* and Sandrine Baghdoyan 2 1 CECS/AFM, I-STEM, 91100 Corbeil-Essonnes, France; [email protected] (L.B.); [email protected] (J.G.); [email protected] (T.Q.); [email protected] (M.P.) 2 INSERM/ UEVE UMR 861, Paris Saclay Univ I-STEM, 91100 Corbeil-Essonnes, France; [email protected] (L.L.); [email protected] (J.A.D.); [email protected] (M.C.); [email protected] (L.A.); [email protected] (S.B.) 3 LEE/ CNRGH/ CEA—IBFJ 2, 91000 Evry, France; [email protected] * Correspondence: [email protected] Received: 27 August 2019; Accepted: 19 November 2019; Published: 26 November 2019 Abstract: Substantial variations in differentiation properties have been reported among human pluripotent cell lines (hPSC), which could affect their utility and clinical safety. We characterized the variable osteogenic capacity observed between different human pluripotent stem cell lines. By focusing on the miRNA expression profile, we demonstrated that the osteogenic differentiation propensity of human pluripotent stem cell lines could be associated with the methylation status and the expression of miRNAs from the imprinted DLK1/DIO3 locus. More specifically, quantitative analysis of the expression of six different miRNAs of that locus prospectively identified human embryonic stem cells and human-induced pluripotent stem cells with differential osteogenic differentiation capacities. -

The Application of Tissue Engineering to Regeneration of Pulp and Dentin in Endodontics Misako Nakashima, Phd, DDS, and Akifumi Akamine, Phd, DDS
Review The Application of Tissue Engineering to Regeneration of Pulp and Dentin in Endodontics Misako Nakashima, PhD, DDS, and Akifumi Akamine, PhD, DDS Abstract Caries, pulpitis, and apical periodontitis increase health here is a high rate of success in retention of teeth by endodontic therapy. A recent care costs and attendant loss of economic productivity. Tstudy of more than 1.4 million cases indicate that about 97% of treated teeth remain They ultimately result in premature tooth loss and functional over an 8-yr follow-up period (1). However, many teeth are not restorable therefore diminishing the quality of life. Advances in because of apical resorption and fracture, incompletely formed roots, or carious de- vital pulp therapy with pulp stem/progenitor cells might struction of coronal structures. In addition, vital pulp therapy is not always predictable. give impetus to regenerate dentin-pulp complex with- One novel approach to restore tooth structure is based on biology: regenerative end- out the removal of the whole pulp. Tissue engineering odontic procedures by application of tissue engineering. Over the last two decades, is the science of design and manufacture of new tissues tissue engineering has evolved from science fiction to science. Indeed, isolated clinical to replace lost parts because of diseases including case reports are consistent with the concept that certain clinical treatments might evolve cancer and trauma. The three key ingredients for tissue into regenerative endodontic procedures (2). However, additional translational re- engineering are signals for morphogenesis, stem cells search is needed to develop predictable clinical regenerative procedures. The purpose for responding to morphogens and the scaffold of of this article is to review the biological principles of tissue engineering and the hurdles extracellular matrix. -

Context-Dependent Roles in Cell and Tissue Physiology
Downloaded from http://cshperspectives.cshlp.org/ on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press TGF-b and the TGF-b Family: Context-Dependent Roles in Cell and Tissue Physiology Masato Morikawa,1 Rik Derynck,2 and Kohei Miyazono3 1Ludwig Cancer Research, Science for Life Laboratory, Uppsala University, Biomedical Center, SE-751 24 Uppsala, Sweden 2Department of Cell and Tissue Biology, University of California at San Francisco, San Francisco, California 94143 3Department of Molecular Pathology, Graduate School of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan Correspondence: [email protected] The transforming growth factor-b (TGF-b) is the prototype of the TGF-b family of growth and differentiation factors, which is encoded by 33 genes in mammals and comprises homo- and heterodimers. This review introduces the reader to the TGF-b family with its complexity of names and biological activities. It also introduces TGF-b as the best-studied factor among the TGF-b family proteins, with its diversity of roles in the control of cell proliferation and differentiation, wound healing and immune system, and its key roles in pathology, for exam- ple, skeletal diseases, fibrosis, and cancer. lthough initially thought to stimulate cell TGF-b has been well documented in most cell Aproliferation, just like many growth factors, types, and has been best characterized in epithe- it became rapidly accepted that transforming lial cells. The bifunctional and context-depen- growth factor b (TGF-b) is a bifunctional reg- dent nature of TGF-b activities was further con- ulator that either inhibits or stimulates cell pro- firmed in a large variety of cell systems and liferation. -

Latent TGF-Β Structure and Activation
ARTICLE doi:10.1038/nature10152 Latent TGF-b structure and activation Minlong Shi1, Jianghai Zhu1, Rui Wang1,XingChen1, Lizhi Mi1, Thomas Walz2 & Timothy A. Springer1 Transforming growth factor (TGF)-b is stored in the extracellular matrix as a latent complex with its prodomain. Activation of TGF-b1 requires the binding of av integrin to an RGD sequence in the prodomain and exertion of force on this domain, which is held in the extracellular matrix by latent TGF-b binding proteins. Crystals of dimeric porcine proTGF-b1 reveal a ring-shaped complex, a novel fold for the prodomain, and show how the prodomain shields the growth factor from recognition by receptors and alters its conformation. Complex formation between avb6 integrin and the prodomain is insufficient for TGF-b1 release. Force-dependent activation requires unfastening of a ‘straitjacket’ that encircles each growth-factor monomer at a position that can be locked by a disulphide bond. Sequences of all 33 TGF-b family members indicate a similar prodomain fold. The structure provides insights into the regulation of a family of growth and differentiation factors of fundamental importance in morphogenesis and homeostasis. The TGF-b family is key to specifying the body plan during metazoan Crystal structure development1,2. Members of this family, including nodal, activins, The structure of pro-TGF-b1 at 3.05 A˚ (Fig. 1a–c and Supplementary inhibins, bone morphogenetic proteins (BMPs) and growth differ- Table 1) was solved using multi- and single-wavelength anomalous entiation factors (GDFs), specify the anterior/posterior and dorsal/ diffraction. Electron density maps (Supplementary Fig. -

Primary Chicken and Duck Endothelial Cells Display a Differential Response to Infection with Highly Pathogenic Avian Influenza Virus
G C A T T A C G G C A T genes Article Primary Chicken and Duck Endothelial Cells Display a Differential Response to Infection with Highly Pathogenic Avian Influenza Virus Zhen Wei Marcus Tong 1,†, Anjana C. Karawita 1,2,† , Colin Kern 3, Huaijun Zhou 3 , Jane E. Sinclair 1 , Limin Yan 1, Keng Yih Chew 1, Sue Lowther 2, Lee Trinidad 2, Arjun Challagulla 2 , Karel A. Schat 4 , Michelle L. Baker 2 and Kirsty R. Short 1,5,* 1 School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane 4072, Australia; [email protected] (Z.W.M.T.); [email protected] (A.C.K.); [email protected] (J.E.S.); [email protected] (L.Y.); [email protected] (K.Y.C.) 2 CSIRO, Australian Centre for Disease Preparedness, Health, and Biosecurity Business Unit, Geelong 3219, Australia; [email protected] (S.L.); [email protected] (L.T.); [email protected] (A.C.); [email protected] (M.L.B.) 3 Department of Animal Science, University of California, Davis, CA 95616, USA; [email protected] (C.K.); [email protected] (H.Z.) 4 Department of Microbiology and Immunology, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853, USA; [email protected] 5 Australian Infectious Diseases Research Centre, The University of Queensland, Brisbane 4072, Australia * Correspondence: [email protected] † These authors contributed equally to this work. Citation: Tong, Z.W.M.; Karawita, A.C.; Kern, C.; Zhou, H.; Sinclair, J.E.; Abstract: Highly pathogenic avian influenza viruses (HPAIVs) in gallinaceous poultry are associated Yan, L.; Chew, K.Y.; Lowther, S.; Trinidad, L.; Challagulla, A.; et al. -

Interplay Between Electrical Activity and Bone Morphogenetic Protein Signaling Regulates Spinal Neuron Differentiation
Interplay between electrical activity and bone morphogenetic protein signaling regulates spinal neuron differentiation Immani Swapna1 and Laura N. Borodinsky2 Department of Physiology and Membrane Biology, Shriners Hospital for Children Northern California, University of California at Davis School of Medicine, Sacramento, CA 95817 Edited by Lynn T. Landmesser, Case Western Reserve University, Cleveland, OH, and approved August 28, 2012 (received for review February 17, 2012) A gradient of bone morphogenetic proteins (BMPs) along the Concurrently, while neurons are being generated and special- + dorsoventral axis of the spinal cord is necessary for the specification ized, they exhibit spontaneous Ca2 -mediated electrical activity of dorsal neurons. Concurrently, a gradient of calcium-mediated (15). This activity occurs prior and during synapse formation and electrical activity is present in the developing spinal cord but in an modulates several aspects of nervous system development. It is opposing ventrodorsal direction. Whether BMPs and electrical important for the proliferation of mouse cortical progenitors (16), activity interact in embryonic spinal neurons remains unknown. cell migration of cerebellar, hippocampal, and cortical cells in We show that BMP decreases electrical activity by enhancing p38 mice (17–19), differentiation of spinal and cerebral neurons in MAPK-mediated negative modulation of voltage-gated sodium Xenopus and mouse (20–25), and pathfinding of retinogeniculate channels. In turn, electrical activity affects the phosphorylation projections in ferret and of motor neuron axons in chicken (26, status and nuclear level of activated Smads, the canonical compo- 27), demonstrating the universality of the relevance of early nents of BMP signaling. This interaction between calcium spike electrical activity for nervous system development. -

Multi-Omics Analysis Reveals the Pan-Cancer Landscape of Bone Morphogenetic Proteins
DATABASE ANALYSIS e-ISSN 1643-3750 © Med Sci Monit, 2020; 26: e920943 DOI: 10.12659/MSM.920943 Received: 2019.10.24 Accepted: 2020.01.27 Multi-Omics Analysis Reveals the Pan-Cancer Available online: 2020.02.12 Published: 2020.04.05 Landscape of Bone Morphogenetic Proteins Authors’ Contribution: AB Wen-Li Luo Department of Orthopedics, Ningbo Hangzhou Bay Hospital, Ningbo, Zhejiang, Study Design A ABCDEFG Ming-Xing Luo P.R. China Data Collection B Statistical Analysis C ABCD Rong-Zhen He Data Interpretation D ACD Lv-Fang Ying Manuscript Preparation E A Jian Luo Literature Search F Funds Collection G Corresponding Author: Ming-Xing Luo, e-mail: [email protected], [email protected] Source of support: Departmental sources Background: Bone morphogenetic proteins (BMPs) are widely involved in cancer development. However, a wealth of con- flicting data raises the question of whether BMPs serve as oncogenes or as cancer suppressors. Material/Methods: By integrating multi-omics data across cancers, we comprehensively analyzed the genomic and pharmacoge- nomic landscape of BMP genes across cancers. Results: Surprisingly, our data indicate that BMPs are globally downregulated in cancers. Further genetics and epi- genetics analyses show that this abnormal expression is driven by copy number variations, especially hetero- zygous amplification. We next assessed the BMP-associated pathways and demonstrated that they suppress cell cycle and estrogen hormone pathways. Bone morphogenetic protein interacts with 58 compounds, and their dysfunction can induce drug sensitivity. Conclusions: Our results define the landscape of the BMP family at a systems level and open potential therapeutic oppor- tunities for cancer patients. -
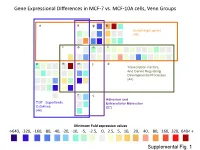
Supplementary Data
Gene Expressional Differences in MCF-7 vs. MCF-10A cells, Venn Groups e k g b Smad target genes (48) l o n i a h m j d Transcription Factors, And Genes Regulating Developmental Processes (44) f c Adhesion and TGF Superfamily Extracellular Molecules Cytokines (27) (44) Minimum Fold expression values ->640, -320, -160, -80, -40, -20, -10, -5, -2.5, 0, 2.5, 5, 10, 20, 40, 80, 160, 320, 640< + Supplemental Fig. 1 Supplemental Table 1. Change of Basal Gene Expressional values in MCF-7 as compared to MCF-10A cell line. Fold T-test Gene # GeneBank Symbol Up/Down p-Value Description /Position Regulation Venn Group a 01 /A01 NM_001105 ACVR1 -4.96 0.0000 Activin A receptor, type I 02 /A02 NM_001616 ACVR2A -2.62 0.0007 Activin A receptor, type IIA 03 /A03 NM_000020 ACVRL1 -1.25 0.7013 Activin A receptor type II-like 1 05 /A05 NM_020547 AMHR2 1.08 0.8043 Anti-Mullerian hormone receptor, type II 16 /B04 NM_004329 BMPR1A -2.40 0.0011 Bone morphogenetic protein receptor, type IA 17 /B05 NM_001203 BMPR1B -7.37 0.0000 Bone morphogenetic protein receptor, type IB 36 /C12 NM_000557 GDF5 -1.38 0.4911 Growth differentiation factor 5 (cartilage-derived morphogenetic protein-1) 37 /D01 NM_001001557 GDF6 1.12 0.9002 Growth differentiation factor 6 38 /D02 NM_182828 GDF7 -1.52 0.4995 Growth differentiation factor 7 53 /E05 NM_020997 LEFTY1 -1.76 0.0529 Left-right determination factor 1 59 /E11 NM_018055 NODAL -3.62 0.1290 Nodal homolog (mouse) 77 /G05 NM_003238 TGFB2 -4.52 0.0566 Transforming growth factor, beta 2 78 /G06 NM_003239 TGFB3 -1.12 0.2902 Transforming -

CER1 Is a Common Target of WNT and NODAL Signaling Pathways in Human Embryonic Stem Cells
795-799 24/3/06 13:04 Page 795 INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 17: 795-799, 2006 795 CER1 is a common target of WNT and NODAL signaling pathways in human embryonic stem cells MASUKO KATOH1 and MASARU KATOH2 1M&M Medical BioInformatics, Hongo 113-0033; 2Genetics and Cell Biology Section, National Cancer Center Research Institute, Tokyo 104-0045, Japan Received January 3, 2006; Accepted February 7, 2006 Abstract. Nodal and BMP signaling pathways network with embryogenesis, underwent protein evolution as well as WNT signaling pathway during embryogenesis and carcino- promoter evolution. These facts indicate that molecular genesis. CER1 (Cerberus 1) and GREM3 (CKTSF1B3 or evolution of CER1 orthologs contributes to the significantly CER2) inhibit NODAL signaling through ACVR1B (ALK4) divergent scenarios of early embryogenesis in primates and or ACVR1C (ALK7) to SMAD2 or SMAD3. GREM1 rodents. (CKTSF1B1) inhibits BMP signaling through BMPR1A (ALK3), BMPR1B (ALK6) or ACVR1 (ALK2) to SMAD1, Introduction SMAD5 or SMAD8. CER1, GREM1 and GREM3 are DAN domain (DAND) family members; however, transcriptional TGFB1, TGFB2, TGFB3, NODAL, LEFTY1, LEFTY2, INHA, regulation of DAND family members by canonical WNT INHBA, INHBB, INHBC, INHBE, AMH, BMP2, BMP3, signaling pathway remains unclear. We searched for the BMP4, BMP5, BMP6, BMP7, BMP8A, BMP8B, BMP10, TCF/LEF-binding site within the promoter region of DAND BMP15, GDF1, GDF2, GDF3, GDF5, GDF6, GDF7, GDF8, family genes, including CER1, GREM1, GREM2, GREM3 and GDF9, GDF10, GDF11, and GDF15 are TGFß superfamily NBL1. Because triple TCF/LEF-binding sites were identified genes within the human genome (http://www.gene.ucl.ac.uk). within human CER1 promoter by using bioinformatics and TGFß signals are transduced through type I receptor TGFBR1 human intelligence, comparative genomics analyses on CER1 and type II receptor TGFBR2 to phosphorylate R-SMAD orthologs were further performed.