Downloaded Existing Trichoptera Genomes from Genbank
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Trait-Based Approach to Assess the Vulnerability of European Aquatic Insects to Climate Change
Hydrobiologia (2014) 721:297–315 DOI 10.1007/s10750-013-1690-7 PRIMARY RESEARCH PAPER A trait-based approach to assess the vulnerability of European aquatic insects to climate change Lorenza Conti • Astrid Schmidt-Kloiber • Gae¨l Grenouillet • Wolfram Graf Received: 15 May 2013 / Revised: 9 August 2013 / Accepted: 14 September 2013 / Published online: 27 September 2013 Ó Springer Science+Business Media Dordrecht 2013 Abstract Aquatic insects are the dominant taxon ecoregions. By means of a fuzzy correspondence group in most freshwater ecosystems. As temperature analysis (FCA), two gradients emerged: (1) a longi- is the main driver of their life cycle development, tudinal gradient, describing successive upstream– metabolic activity, and geographic distribution, these downstream features, and (2) a biogeographical gra- macroinvertebrates are particularly suitable for large dient, separating endemic and micro-endemic species scale and comparative studies of freshwater commu- from widely distributed taxa. Moreover, aquatic nity responses to climate change. A dataset of bio- insects of southern European ecoregions emerged as ecological traits of 1,942 Ephemeroptera, Plecoptera, those most endangered in terms of potential vulner- and Trichoptera (EPT) taxa was used to analyze (1) the ability to climate change. Comparative multi-taxon relationships among traits, (2) the potential vulnera- studies provide important new insights into freshwater bility of EPT species to climate change, and (3) the ecosystem functioning and responses to climate geographical occurrence patterns of these potentially change, and could be the first step toward developing endangered species at the scale of European integrative monitoring or assessment tools (e.g., trait- based indicators at the species level) by means of non- arbitrary statistical methods. -

Research Article
Ecologica Montenegrina 44: 69-95 (2021) This journal is available online at: www.biotaxa.org/em http://dx.doi.org/10.37828/em.2021.44.10 Biodiversity, DNA barcoding data and ecological traits of caddisflies (Insecta, Trichoptera) in the catchment area of the Mediterranean karst River Cetina (Croatia) IVAN VUČKOVIĆ1*, MLADEN KUČINIĆ2**, ANĐELA ĆUKUŠIĆ3, MARIJANA VUKOVIĆ4, RENATA ĆUK5, SVJETLANA STANIĆ-KOŠTROMAN6, DARKO CERJANEC7 & MLADEN PLANTAK1 1Elektroprojekt d.d., Civil and Architectural Engineering Department, Section of Ecology, Alexandera von Humboldta 4, 10 000 Zagreb, Croatia. E-mails:[email protected]; [email protected] 2Department of Biology (Laboratory for Entomology), Faculty of Science, University of Zagreb, Rooseveltov trg 6, 10 000 Zagreb, Croatia. E-mail: [email protected] 3Ministry of Economy and Sustainable Development, Radnička cesta 80/7, 10000 Zagreb, Croatia. E-mail: [email protected] 4Croatian Natural History Museum, Demetrova 1, 10 000 Zagreb, Croatia. E-mail: [email protected] 5Hrvatske vode, Central Water Management Laboratory, Ulica grada Vukovara 220, 10 000 Zagreb, Croatia. E-mail:[email protected] 6Faculty of Science and Education, University of Mostar, Matice hrvatske bb, 88000 Mostar, Bosnia and Herzegovina. E-mail: [email protected] 7Primary School Barilović, Barilović 96, 47252 Barilović and Primary School Netretić, Netretić 1, 47271 E-mail: [email protected] *Corresponding author: [email protected] **Equally contributing author Received 2 June 2021 │ Accepted by V. Pešić: 19 July 2021 │ Published online 2 August 2021. Abstract The environmental and faunistic research conducted included defining the composition and distribution of caddisflies collected using ultraviolet (UV) light trap at 11 stations along the Cetina River, from the spring to the mouth, and also along its tributaries the Ruda River and the Grab River with two sampling stations each, and the Rumin River with one station. -
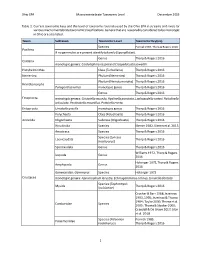
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 1 Table 1. Current Taxonomic Keys and the Level of Taxonomy Routinely U
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Table 1. Current taxonomic keys and the level of taxonomy routinely used by the Ohio EPA in streams and rivers for various macroinvertebrate taxonomic classifications. Genera that are reasonably considered to be monotypic in Ohio are also listed. Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Species Pennak 1989, Thorp & Rogers 2016 Porifera If no gemmules are present identify to family (Spongillidae). Genus Thorp & Rogers 2016 Cnidaria monotypic genera: Cordylophora caspia and Craspedacusta sowerbii Platyhelminthes Class (Turbellaria) Thorp & Rogers 2016 Nemertea Phylum (Nemertea) Thorp & Rogers 2016 Phylum (Nematomorpha) Thorp & Rogers 2016 Nematomorpha Paragordius varius monotypic genus Thorp & Rogers 2016 Genus Thorp & Rogers 2016 Ectoprocta monotypic genera: Cristatella mucedo, Hyalinella punctata, Lophopodella carteri, Paludicella articulata, Pectinatella magnifica, Pottsiella erecta Entoprocta Urnatella gracilis monotypic genus Thorp & Rogers 2016 Polychaeta Class (Polychaeta) Thorp & Rogers 2016 Annelida Oligochaeta Subclass (Oligochaeta) Thorp & Rogers 2016 Hirudinida Species Klemm 1982, Klemm et al. 2015 Anostraca Species Thorp & Rogers 2016 Species (Lynceus Laevicaudata Thorp & Rogers 2016 brachyurus) Spinicaudata Genus Thorp & Rogers 2016 Williams 1972, Thorp & Rogers Isopoda Genus 2016 Holsinger 1972, Thorp & Rogers Amphipoda Genus 2016 Gammaridae: Gammarus Species Holsinger 1972 Crustacea monotypic genera: Apocorophium lacustre, Echinogammarus ischnus, Synurella dentata Species (Taphromysis Mysida Thorp & Rogers 2016 louisianae) Crocker & Barr 1968; Jezerinac 1993, 1995; Jezerinac & Thoma 1984; Taylor 2000; Thoma et al. Cambaridae Species 2005; Thoma & Stocker 2009; Crandall & De Grave 2017; Glon et al. 2018 Species (Palaemon Pennak 1989, Palaemonidae kadiakensis) Thorp & Rogers 2016 1 Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Informal grouping of the Arachnida Hydrachnidia Smith 2001 water mites Genus Morse et al. -

Understanding Brassicaceae Evolution Through Ancestral
Understanding Brassicaceae evolution through ancestral genome reconstruction Florent Murat, Alexandra Louis, Florian Maumus, Alix Armero Villanueva, Richard Cooke, Hadi Quesneville, Hugues Roest Crollius, Jerome Salse To cite this version: Florent Murat, Alexandra Louis, Florian Maumus, Alix Armero Villanueva, Richard Cooke, et al.. Understanding Brassicaceae evolution through ancestral genome reconstruction. Genome Biology, BioMed Central, 2015, 16 (262), 10.1186/s13059-015-0814-y. hal-01281832 HAL Id: hal-01281832 https://hal.archives-ouvertes.fr/hal-01281832 Submitted on 2 Mar 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Murat et al. Genome Biology (2015) 16:262 DOI 10.1186/s13059-015-0814-y RESEARCH Open Access Understanding Brassicaceae evolution through ancestral genome reconstruction Florent Murat1†, Alexandra Louis2,3,4†, Florian Maumus5†, Alix Armero1, Richard Cooke6, Hadi Quesneville5, Hugues Roest Crollius2,3,4 and Jerome Salse1* Abstract Background: Brassicaceae is a family of green plants of high scientific and economic interest, including thale cress (Arabidopsis thaliana), cruciferous vegetables (cabbages) and rapeseed. Results: We reconstruct an evolutionary framework of Brassicaceae composed of high-resolution ancestral karyotypes using the genomes of modern A. thaliana, Arabidopsis lyrata, Capsella rubella, Brassica rapa and Thellungiella parvula. -

Developmental Trade-Offs and Resource Allocation in Caddis Flies
Stevens, David J. (2000) Developmental trade-offs and resource allocation in caddis flies. PhD thesis http://theses.gla.ac.uk/3764/ Copyright and moral rights for this thesis are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the Author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the Author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Glasgow Theses Service http://theses.gla.ac.uk/ [email protected] Developmental Trade-Offs and Resource Allocation In Caddis Flies. David J. Stevens A thesis submitted for the degree ofDoctor ofPhilosophy to the Faculty of Science, University ofGlasgow. September 2000 Declaration I declare that the work presented in this thesis has been completed by myselfunless otherwise acknowledged in the text. No part ofthis work has been presented for any other qualification. David Stevens September 2000 Acknowledgements I could not have hoped for two better supervisors than Pat Monaghan and Mike Hansell. I wish to thank them not just for giving me my chance in the first place, but for giving me the freedom within the project to explore many avenues ofinvestigation. Most ofthese proved fruitful, ifonly to teach me not to do it again. The support, encouragement and advice they gave were ofimmeasurable help over the course ofthe project. -

Dna Barcodes, Partitioned Phylogenetic Models, And
LARGE DATASETS AND TRICHOPTERA PHYLOGENETICS: DNA BARCODES, PARTITIONED PHYLOGENETIC MODELS, AND THE EVOLUTION OF PHRYGANEIDAE By PAUL BRYAN FRANDSEN A dissertation submitted to the Graduate School-New Brunswick Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Entomology Written under the direction of Karl M. Kjer And approved by _____________________________________ _____________________________________ _____________________________________ _____________________________________ New Brunswick, New Jersey OCTOBER 2015 ABSTRACT OF THE DISSERTATION Large datasets and Trichoptera phylogenetics: DNA barcodes, partitioned phylogenetic models, and the evolution of Phryganeidae By PAUL BRYAN FRANDSEN Dissertation Director: Karl M. Kjer Large datasets in phylogenetics—those with a large number of taxa, e.g. DNA barcode data sets, and those with a large amount of sequence data per taxon, e.g. data sets generated from high throughput sequencing—pose both exciting possibilities and interesting analytical problems. The analysis of both types of large datasets is explored in this dissertation. First, the use of DNA barcodes in phylogenetics is investigated via the generation of phylogenetic trees for known monophyletic clades. Barcodes are found to be useful in shallow scale phylogenetic analyses when given a well-supported scaffold on which to place them. One of the analytical challenges posed by large phylogenetic datasets is the selection of appropriate partitioned models of molecular evolution. The most commonly used model partitioning strategies can fail to characterize the true variation of the evolutionary process and this effect can be exacerbated when applied to large datasets. A new, scalable algorithm for the automatic selection ! ii! of partitioned models of molecular evolution is proposed with an eye toward reducing systematic error in phylogenomics. -

The Future of Transposable Element Annotation and Their Classification in the Light of Functional Genomics
The future of transposable element annotation and their classification in the light of functional genomics -what we can learn from the fables of Jean de la Fontaine? Peter Arensburger, Benoit Piegu, Yves Bigot To cite this version: Peter Arensburger, Benoit Piegu, Yves Bigot. The future of transposable element annotation and their classification in the light of functional genomics - what we can learn from thefables of Jean de la Fontaine?. Mobile Genetic Elements, Taylor & Francis, 2016, 6 (6), pp.e1256852. 10.1080/2159256X.2016.1256852. hal-02385838 HAL Id: hal-02385838 https://hal.archives-ouvertes.fr/hal-02385838 Submitted on 27 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Copyright The future of transposable element annotation and their classification in the light of functional genomics - what we can learn from the fables of Jean de la Fontaine? Peter Arensburger1, Benoît Piégu2, and Yves Bigot2 1 Biological Sciences Department, California State Polytechnic University, Pomona, CA 91768 - United States of America. 2 Physiologie de la reproduction et des Comportements, UMR INRA-CNRS 7247, PRC, 37380 Nouzilly – France Corresponding author address: Biological Sciences Department, California State Polytechnic University, Pomona, CA 91768 - United States of America. -

Marine Insects
UC San Diego Scripps Institution of Oceanography Technical Report Title Marine Insects Permalink https://escholarship.org/uc/item/1pm1485b Author Cheng, Lanna Publication Date 1976 eScholarship.org Powered by the California Digital Library University of California Marine Insects Edited by LannaCheng Scripps Institution of Oceanography, University of California, La Jolla, Calif. 92093, U.S.A. NORTH-HOLLANDPUBLISHINGCOMPANAY, AMSTERDAM- OXFORD AMERICANELSEVIERPUBLISHINGCOMPANY , NEWYORK © North-Holland Publishing Company - 1976 All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise,without the prior permission of the copyright owner. North-Holland ISBN: 0 7204 0581 5 American Elsevier ISBN: 0444 11213 8 PUBLISHERS: NORTH-HOLLAND PUBLISHING COMPANY - AMSTERDAM NORTH-HOLLAND PUBLISHING COMPANY LTD. - OXFORD SOLEDISTRIBUTORSFORTHEU.S.A.ANDCANADA: AMERICAN ELSEVIER PUBLISHING COMPANY, INC . 52 VANDERBILT AVENUE, NEW YORK, N.Y. 10017 Library of Congress Cataloging in Publication Data Main entry under title: Marine insects. Includes indexes. 1. Insects, Marine. I. Cheng, Lanna. QL463.M25 595.700902 76-17123 ISBN 0-444-11213-8 Preface In a book of this kind, it would be difficult to achieve a uniform treatment for each of the groups of insects discussed. The contents of each chapter generally reflect the special interests of the contributors. Some have presented a detailed taxonomic review of the families concerned; some have referred the readers to standard taxonomic works, in view of the breadth and complexity of the subject concerned, and have concentrated on ecological or physiological aspects; others have chosen to review insects of a specific set of habitats. -

The Zoogeomorphology of Case-Building Caddisfly Larvae
The zoogeomorphology of case-building caddisfly larvae by Richard Mason A Doctoral thesis submitted in partial fulfilment of the requirements for the award of Doctor of Philosophy of Loughborough University (June 2020) © Richard Mason 2020 i Abstract Caddisfly (Trichoptera) are an abundant and widespread aquatic insect group. Caddisfly larvae of most species build cases from silk and fine sediment at some point in their lifecycle. Case- building caddisfly have the potential to modify the distribution and transport of sediment by: 1) altering sediment properties through case construction, and 2) transporting sediment incorporated into cases over the riverbed. This thesis investigates, for the first time, the effects of bioconstruction by case-building caddisfly on fluvial geomorphology. The research was conducted using two flume experiments to understand the mechanisms of caddisfly zoogeomorphology (case construction and transporting sediment), and two field investigations that increase the spatial and temporal scale of the research. Caddisfly cases varied considerably in mass between species (0.001 g - 0.83 g) and grain sizes used (D50 = 0.17 mm - 4 mm). As a community, caddisfly used a wide range of grain-sizes in case construction (0.063 mm – 11 mm), and, on average, the mass of incorporated sediment was 38 g m-2, in a gravel-bed stream. This sediment was aggregated into biogenic particles (cases) which differed in size and shape from their constituent grains. A flume experiment determined that empty cases of some caddisfly species (tubular case-builders; Limnephilidae and Sericostomatidae) were more mobile than their incorporated sediment, but that dome shaped Glossosomatidae cases moved at the same entrainment threshold as their constituent grains, highlighting the importance of case design as a control on caddisfly zoogeomorphology. -
New Species and a New Genus of Philopotamidae from the Andes of Bolivia and Ecuador (Insecta, Trichoptera)
A peer-reviewed open-access journal ZooKeys New780: 89–108 species (2018) and a new genus of Philopotamidae from the Andes of Bolivia and Ecuador... 89 doi: 10.3897/zookeys.780.26977 RESEARCH ARTICLE http://zookeys.pensoft.net Launched to accelerate biodiversity research New species and a new genus of Philopotamidae from the Andes of Bolivia and Ecuador (Insecta, Trichoptera) Ralph W. Holzenthal1, Roger J. Blahnik1, Blanca Ríos-Touma2,3 1 Department of Entomology, University of Minnesota, 1980 Folwell Avenue, 219 Hodson Hall, St. Paul, Minnesota 55108 USA 2 Facultad de Ingenierías y Ciencias Aplicadas, Ingeniería Ambiental, Grupo de In- vestigación en Biodiversidad Medio Ambiente y Salud – BIOMAS – Universidad de Las Américas, Campus Queri, Quito, Ecuador 3 Instituto Nacional de Biodiversidad, Quito, Pichincha, Ecuador Corresponding author: Ralph Holzenthal ([email protected]) Academic editor: S. Vitecek | Received 25 May 2018 | Accepted 19 July 2018 | Published 8 August 2018 http://zoobank.org/04DB004E-E4F9-4B94-8EC8-37656481D190 Citation: Holzenthal RW, Blahnik RJ, Ríos-Touma B (2018) New species and a new genus of Philopotamidae from the Andes of Bolivia and Ecuador (Insecta, Trichoptera). ZooKeys 780: 89–108. https://doi.org/10.3897/zookeys.780.26977 Abstract A new genus and species of Philopotamidae (Philopotaminae), Aymaradella boliviana, is described from the Bolivian Andes of South America. The new genus differs from other Philopotaminae by the loss of 2A vein in the hind wing and, in the male genitalia, the synscleritous tergum and sternum of seg- ment VIII, and the elongate sclerotized dorsal processes of segment VIII. The first record ofHydrobiosella (Philopotaminae) in the New World is also provided with a new species from the Andes of Ecuador, Hydrobiosella andina. -

1 New Ohio and Indiana Records of Aquatic Insects (Ephemeroptera
Ohio Biological Survey Notes 9: 1–15, 2019. © Ohio Biological Survey, Inc. New Ohio and Indiana Records of Aquatic Insects (Ephemeroptera, Plecoptera, Trichoptera, Coleoptera: Elmidae, Diptera: Chironomidae) MICHAEL J. BOLTON1, SARAH K. MACY2, R. EDWARD DEWALT3, AND LUKE M. JACOBUS4 1Ohio Environmental Protection Agency, Division of Surface Water, 4675 Homer Ohio Lane, Groveport, OH 43125, Michael.Bolton@epa. ohio.gov; 2Formerly with the Ohio Environmental Protection Agency; current e-mail: [email protected]; 3University of Illinois, Illinois Natural History Survey, 1816 S Oak St., Champaign, IL 61820, [email protected]; 4Indiana University–Purdue University Columbus, 4601 Central Avenue, Columbus, IN 47203, [email protected]. Abstract: New state records and additional locations for rarely collected species are reported for Ephemeroptera (mayflies), Plecoptera (stoneflies), Trichoptera (caddisflies), Coleoptera: Elmidae (riffle beetles), and Diptera: Chironomidae (chironomids, non-biting midges, midges). These specimen records result primarily from Ohio Environmental Protection Agency biomonitoring of Ohio streams and from records found in the Purdue University Entomological Research Collection and the Illinois Natural History Survey Insect Collection; a few records were derived from material housed in two other collections. New state records for Ohio consist of the mayflies Acentrella rallatoma Burian & Myers, Acerpenna pygmaea (Hagen), Anafroptilum album (McDunnough), Anafroptilum minor group species 1, Anafroptilum -

Of the Korean Peninsula
Journal288 of Species Research 9(3):288-323, 2020JOURNAL OF SPECIES RESEARCH Vol. 9, No. 3 A checklist of Trichoptera (Insecta) of the Korean Peninsula Sun-Jin Park and Dongsoo Kong* Department of Life Science, Kyonggi University, Suwon 16227, Republic of Korea *Correspondent: [email protected] A revised checklist of Korean Trichoptera is provided for the species recorded from the Korean Peninsula, including both North and South Korea. The checklist includes bibliographic research as well as results after reexamination of some specimens. For each species, we provide the taxonomic literature that examined Korean Trichoptera materials or mentioned significant taxonomic treatments regarding to Korean species. We also provide the records of unnamed species based on larval identification for further study. Based on taxonomic considerations, 20 species among the previously known nominal species in Korea are deleted or synonymized, and three species omitted from the previous lists, Hydropsyche athene Malicky and Chantaramongkol, 2000, H. simulata Mosely, 1942 and Helicopsyche coreana Mey, 1991 are newly added to the checklist. Hydropsyche formosana Ulmer, 1911 is recorded from the Korean Peninsula for the first time by the identification of Hydropsyche KD. In addition, we recognized 14 species of larvae separated with only tentative alphabetic designations. As a result, this new Korean Trichoptera checklist includes 218 currently recognized species in 66 genera and 25 families from the Korean Peninsula. Keywords: caddisflies, catalogue, history, North Korea, South Korea Ⓒ 2020 National Institute of Biological Resources DOI:10.12651/JSR.2020.9.3.288 INTRODUCTION Democratic Republic (North Korea). Since the mid 1970s, several scientists within the Republic of Korea (South Trichoptera is the seventh-largest order among Insecta, Korea) have studied Trichoptera.