Dna Barcodes, Partitioned Phylogenetic Models, And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

USDA, Forest Service Forest Health Protection GSA Contract No
SERA TR 02-43-13-03b Triclopyr - Revised Human Health and Ecological Risk Assessments Final Report Prepared for: USDA, Forest Service Forest Health Protection GSA Contract No. GS-10F-0082F USDA Forest Service BPA: WO-01-3187-0150 USDA Purchase Order No.: 43-1387-2-0245 Task No. 13 Submitted to: Dave Thomas, COTR Forest Health Protection Staff USDA Forest Service Rosslyn Plaza Building C, Room 7129C 1601 North Kent Street Arlington, VA 22209 Submitted by: Patrick R. Durkin Syracuse Environmental Research Associates, Inc. 5100 Highbridge St., 42C Fayetteville, New York 13066-0950 Telephone: (315) 637-9560 Fax: (315) 637-0445 E-Mail: [email protected] Home Page: www.sera-inc.com March 15, 2003 TABLE OF CONTENTS LIST OF APPENDICES ...................................................... iv LIST OF WORKSHEETS ...................................................... v LIST OF ATTACHMENTS .................................................... v LIST OF TABLES ............................................................ v LIST OF FIGURES ......................................................... viii ACRONYMS, ABBREVIATIONS, AND SYMBOLS .............................. ix COMMON UNIT CONVERSIONS AND ABBREVIATIONS ......................... xi CONVERSION OF SCIENTIFIC NOTATION .................................... xii EXECUTIVE SUMMARY ................................................... xiii 1. INTRODUCTION ........................................................ 1-1 2. PROGRAM DESCRIPTION ................................................ 2-1 2.1. OVERVIEW -

Using DNA Barcodes to Identify and Classify Living Things
Using DNA Barcodes to Identify and Classify Living Things www.dnabarcoding101.org LABORATORY Using DNA Barcodes to Identify and Classify Living Things OBJECTIVES This laboratory demonstrates several important concepts of modern biology. During this laboratory, you will: • Collect and analyze sequence data from plants, fungi, or animals—or products made from them. • Use DNA sequence to identify species. • Explore relationships between species. In addition, this laboratory utilizes several experimental and bioinformatics methods in modern biological research. You will: • Collect plants, fungi, animals, or products in your local environment or neighborhood. • Extract and purify DNA from tissue or processed material. • Amplify a specific region of the chloroplast, mitochondrial, or nuclear genome by polymerase chain reaction (PCR) and analyze PCR products by gel electrophoresis. • Use the Basic Local Alignment Search Tool (BLAST) to identify sequences in databases. • Use multiple sequence alignment and tree-building tools to analyze phylogenetic relation - ships. INTRODUCTION Taxonomy, the science of classifying living things according to shared features, has always been a part of human society. Carl Linneas formalized biological classifi - cation with his system of binomial nomenclature that assigns each organism a genus and species name. Identifying organisms has grown in importance as we monitor the biological effects of global climate change and attempt to preserve species diversity in the face of accelerating habitat destruction. We know very little about the diversity of plants and animals—let alone microbes—living in many unique ecosystems on earth. Less than two million of the estimated 5–50 million plant and animal species have been identified. Scientists agree that the yearly rate of extinction has increased from about one species per million to 100–1,000 species per million. -

DNA Barcoding Distinguishes Pest Species of the Black Fly Genus <I
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications: Department of Entomology Entomology, Department of 11-2013 DNA Barcoding Distinguishes Pest Species of the Black Fly Genus Cnephia (Diptera: Simuliidae) I. M. Confitti University of Toronto K. P. Pruess University of Nebraska-Lincoln A. Cywinska Ingenomics, Inc. T. O. Powers University of Nebraska-Lincoln D. C. Currie University of Toronto and Royal Ontario Museum, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/entomologyfacpub Part of the Entomology Commons Confitti, I. M.; Pruess, K. P.; Cywinska, A.; Powers, T. O.; and Currie, D. C., "DNA Barcoding Distinguishes Pest Species of the Black Fly Genus Cnephia (Diptera: Simuliidae)" (2013). Faculty Publications: Department of Entomology. 616. http://digitalcommons.unl.edu/entomologyfacpub/616 This Article is brought to you for free and open access by the Entomology, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications: Department of Entomology by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. MOLECULAR BIOLOGY/GENOMICS DNA Barcoding Distinguishes Pest Species of the Black Fly Genus Cnephia (Diptera: Simuliidae) 1,2 3 4 5 1,2,6 I. M. CONFLITTI, K. P. PRUESS, A. CYWINSKA, T. O. POWERS, AND D. C. CURRIE J. Med. Entomol. 50(6): 1250Ð1260 (2013); DOI: http://dx.doi.org/10.1603/ME13063 ABSTRACT Accurate species identiÞcation is essential for cost-effective pest control strategies. We tested the utility of COI barcodes for identifying members of the black ßy genus Cnephia Enderlein (Diptera: Simuliidae). Our efforts focus on four Nearctic Cnephia speciesÑCnephia dacotensis (Dyar & Shannon), Cnephia eremities Shewell, Cnephia ornithophilia (Davies, Peterson & Wood), and Cnephia pecuarum (Riley)Ñthe latter two being current or potential targets of biological control programs. -

Species Dossier: Hagenella Clathrata
Species dossier: Hagenella clathrata Window winged sedge July 2011 Mating adult pair Hagenella clathata Contact details Ian Wallace, Curator of Conchology & Aquatic Biology World Museum William Brown Street, Liverpool, L3 8EN Tel: 0151 478 4385 Email: [email protected] Species dossier: Hagenella clathrata Contents Introduction ................................................................................... 3 Summary....................................................................................... 3 Ecology ......................................................................................... 3 History in Britain ............................................................................ 6 European distribution .................................................................... 9 Recent Survey Work ..................................................................... 9 Survey methods ............................................................................ 9 Identification.................................................................................. 9 Threats........................................................................................ 10 Action plan for the Window Winged Sedge ( Hagenella clathrata ) 11 List of references......................................................................... 12 Appendix 2 Records of ( Hagenella clathrata ) from the UK ......... 15 Cover image © Matthew Wallace (2009) Hagenella clathrata (Kolenati, 1848) Window winged sedge (Trichoptera: Phryganeidae) Genus -

List of Animal Species with Ranks October 2017
Washington Natural Heritage Program List of Animal Species with Ranks October 2017 The following list of animals known from Washington is complete for resident and transient vertebrates and several groups of invertebrates, including odonates, branchipods, tiger beetles, butterflies, gastropods, freshwater bivalves and bumble bees. Some species from other groups are included, especially where there are conservation concerns. Among these are the Palouse giant earthworm, a few moths and some of our mayflies and grasshoppers. Currently 857 vertebrate and 1,100 invertebrate taxa are included. Conservation status, in the form of range-wide, national and state ranks are assigned to each taxon. Information on species range and distribution, number of individuals, population trends and threats is collected into a ranking form, analyzed, and used to assign ranks. Ranks are updated periodically, as new information is collected. We welcome new information for any species on our list. Common Name Scientific Name Class Global Rank State Rank State Status Federal Status Northwestern Salamander Ambystoma gracile Amphibia G5 S5 Long-toed Salamander Ambystoma macrodactylum Amphibia G5 S5 Tiger Salamander Ambystoma tigrinum Amphibia G5 S3 Ensatina Ensatina eschscholtzii Amphibia G5 S5 Dunn's Salamander Plethodon dunni Amphibia G4 S3 C Larch Mountain Salamander Plethodon larselli Amphibia G3 S3 S Van Dyke's Salamander Plethodon vandykei Amphibia G3 S3 C Western Red-backed Salamander Plethodon vehiculum Amphibia G5 S5 Rough-skinned Newt Taricha granulosa -

DNA Barcoding Analysis and Phylogenetic Relation of Mangroves in Guangdong Province, China
Article DNA Barcoding Analysis and Phylogenetic Relation of Mangroves in Guangdong Province, China Feng Wu 1,2 , Mei Li 1, Baowen Liao 1,*, Xin Shi 1 and Yong Xu 3,4 1 Key Laboratory of State Forestry Administration on Tropical Forestry Research, Research Institute of Tropical Forestry, Chinese Academy of Forestry, Guangzhou 510520, China; [email protected] (F.W.); [email protected] (M.L.); [email protected] (X.S.) 2 Zhaoqing Xinghu National Wetland Park Management Center, Zhaoqing 526060, China 3 Key Laboratory of Plant Resources Conservation and Sustainable Utilization, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou 510650, China; [email protected] 4 University of Chinese Academy of Sciences, Beijing 100049, China * Correspondence: [email protected]; Tel.: +86-020-8702-8494 Received: 9 December 2018; Accepted: 4 January 2019; Published: 12 January 2019 Abstract: Mangroves are distributed in the transition zone between sea and land, mostly in tropical and subtropical areas. They provide important ecosystem services and are therefore economically valuable. DNA barcoding is a useful tool for species identification and phylogenetic reconstruction. To evaluate the effectiveness of DNA barcoding in identifying mangrove species, we sampled 135 individuals representing 23 species, 22 genera, and 17 families from Zhanjiang, Shenzhen, Huizhou, and Shantou in the Guangdong province, China. We tested the universality of four DNA barcodes, namely rbcL, matK, trnH-psbA, and the internal transcribed spacer of nuclear ribosomal DNA (ITS), and examined their efficacy for species identification and the phylogenetic reconstruction of mangroves. The success rates for PCR amplification of rbcL, matK, trnH-psbA, and ITS were 100%, 80.29% ± 8.48%, 99.38% ± 1.25%, and 97.18% ± 3.25%, respectively, and the rates of DNA sequencing were 100%, 75.04% ± 6.26%, 94.57% ± 5.06%, and 83.35% ± 4.05%, respectively. -

Biographical Sketches
Biological Diversity of the Guiana Shield 2010 Biographical Sketches ADMINISTRATION & BOTANY Christian Feuillet Vicki A. Funk Kaslyn Holder-Collins Carol L. Kelloff Karen Redden Kenneth Wurdack Charles Zartman ECOLOGICAL NICHE MODELING Robert P. Anderson Aleksandar Radosavljevic ENTOMOLOGY Seán G. Brady Matthew L. Buffington Michael William Gates Robert R. Kula John S. LaPolla Ted R. Schultz Jeffrey Sosa-Calvo Jessica Ware ICHTHYOLOGY Calvin Barnard Jonathan W. Armbruster Richard P. Vari HERPETOLOGY Ross D. MacCulloch Brice P. Noonan Robert Reynolds (away from office, no biosketch included) MAMMALS Burton Lim Mark Engstrom (away from office, no biosketch included) ADMINISTRATION & BOTANY Christian Feuillet Vicki A. Funk Kaslyn Holder-Collins Carol L. Kelloff Karen Redden Kenneth Wurdack Charles Zartman Christian P. Feuillet SI-NMNH Research Associate Department of Botany, MRC-166, United States National Herbarium National Museum of Natural History, Smithsonian Institution P.O. Box 37012, Washington, DC 20013-7012 (202) 633-0898 - Fax (202) 786-2563; [email protected] Major Professional Interests / Areas of Expertise Systematics of Gesneriaceae & Passifloraceae and New World Aristolochiaceae & Boraginaceae. Northern South American floristic. Plant architecture. Current Professional Position Research associate in the Department of Botany, Smithsonian Institution. Educational History (France) University of Rouen: Biology–Geology 1971; Chemistry–Biology 1972 University of Science & Technology of Languedoc, Montpellier: Animal Physiology 1973 Mandatory Military Service: Paris, Dec 1973 - Nov 1974 University Paris VI Pierre & Marie Curie, Sobonne: Plant Physiology 1977, Genetics 1977, Botany 1978; Masters in Plant Biology 1978; Ph. D. in Tropical Botany 1981. Career 15 Nov. 1981 – 30 June 1997 Plant taxonomist with ORSTOM (now IRD = Institut de Recherche pour le Développement). -

Publications of Glenn B
Publications: Glenn B. Wiggins, Curator Emeritus, Entomology 2010 Wiggins, G.B. “No small matters. Introducing Biological Notes on an Old Farm: Exploring Common Things in the Kingdoms of Life.” ROM Magazine, 42(2): 29- 31. * 2009 Wiggins, G.B. Biological Notes on an Old Farm: Exploring Common Things in the Kingdoms of Life. Royal Ontario Museum, Toronto. 2008 Wiggins, G.B. and D.C. Currie. “Trichoptera Families.” In An Introduction to the Aquatic Insects of North America, edited by R.W. Merritt, K.W. Cummins, and M.B. Berg. Kendall/Hunt, Dubuque, Iowa (4th edition, revised). 2007 Wiggins, G.B. “Architects under water.” American Entomologist, 53(2): 78-85. 2005b Wiggins, G.B. “Review: Vernal pools, natural history and conservation by Elizabeth A. Colburn.” Journal of the North American Benthological Society, 24(4): 1009-1013. 2005a Vineyard, R.N., G.B. Wiggins, H.E. Frania, and P.W. Schefter. “The caddisfly genus Neophylax (Trichoptera: Uenoidae).” Royal Ontario Museum Contributions in Science, 2: 1-141. * 2004b Wiggins, G.B. Caddisflies: The Underwater Architects. University of Toronto Press. 2004a Wiggins, G.B. “Caddisflies: glimpses into evolutionary history.” Rotunda, 38(2): 32-39. 2002 Wiggins, G.B. “Biogeography of amphipolar caddisflies in the subfamily Dicosmoecinae (Trichoptera, Limnephilidae).” Mitteilungen aus dem Museum für Naturkunde in Berlin, Deutsche Entomologische Zeitschrift, 49(2002) 2: 227- 259. 2001 Wiggins, G.B. “Construction behavior for new pupal cases by case-making caddis larvae: Further comment. (Trichoptera: Integripalpia).” Braueria, 28: 7-9. 1999b Gall, W.K. and G.B. Wiggins. “Evidence bearing on a sister-group relationship between the families Phryganeidae and Plectrotarsidae (Trichoptera).” Proceedings of the Ninth International Symposium on Trichoptera, Chiang Mai, Thailand, 1998, edited by H. -
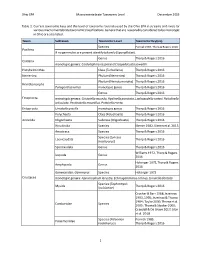
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 1 Table 1. Current Taxonomic Keys and the Level of Taxonomy Routinely U
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Table 1. Current taxonomic keys and the level of taxonomy routinely used by the Ohio EPA in streams and rivers for various macroinvertebrate taxonomic classifications. Genera that are reasonably considered to be monotypic in Ohio are also listed. Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Species Pennak 1989, Thorp & Rogers 2016 Porifera If no gemmules are present identify to family (Spongillidae). Genus Thorp & Rogers 2016 Cnidaria monotypic genera: Cordylophora caspia and Craspedacusta sowerbii Platyhelminthes Class (Turbellaria) Thorp & Rogers 2016 Nemertea Phylum (Nemertea) Thorp & Rogers 2016 Phylum (Nematomorpha) Thorp & Rogers 2016 Nematomorpha Paragordius varius monotypic genus Thorp & Rogers 2016 Genus Thorp & Rogers 2016 Ectoprocta monotypic genera: Cristatella mucedo, Hyalinella punctata, Lophopodella carteri, Paludicella articulata, Pectinatella magnifica, Pottsiella erecta Entoprocta Urnatella gracilis monotypic genus Thorp & Rogers 2016 Polychaeta Class (Polychaeta) Thorp & Rogers 2016 Annelida Oligochaeta Subclass (Oligochaeta) Thorp & Rogers 2016 Hirudinida Species Klemm 1982, Klemm et al. 2015 Anostraca Species Thorp & Rogers 2016 Species (Lynceus Laevicaudata Thorp & Rogers 2016 brachyurus) Spinicaudata Genus Thorp & Rogers 2016 Williams 1972, Thorp & Rogers Isopoda Genus 2016 Holsinger 1972, Thorp & Rogers Amphipoda Genus 2016 Gammaridae: Gammarus Species Holsinger 1972 Crustacea monotypic genera: Apocorophium lacustre, Echinogammarus ischnus, Synurella dentata Species (Taphromysis Mysida Thorp & Rogers 2016 louisianae) Crocker & Barr 1968; Jezerinac 1993, 1995; Jezerinac & Thoma 1984; Taylor 2000; Thoma et al. Cambaridae Species 2005; Thoma & Stocker 2009; Crandall & De Grave 2017; Glon et al. 2018 Species (Palaemon Pennak 1989, Palaemonidae kadiakensis) Thorp & Rogers 2016 1 Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Informal grouping of the Arachnida Hydrachnidia Smith 2001 water mites Genus Morse et al. -

Contribution to the Knowledge of the Caddisfly Fauna (Insecta: Trichoptera) of Leqinat Lakes and Adjacent Streams in Bjeshkët E Nemuna (Kosovo)
View metadata, citation and similar papers at core.ac.uk brought to you by CORE NAT. CROAT. VOL. 28 No 1 35-44 ZAGREB June 30, 2019 original scientific paper / izvorni znanstveni rad DOI 10.20302/NC.2019.28.3 CONTRIBUTION TO THE KNOWLEDGE OF THE CADDISFLY FAUNA (INSECTA: TRICHOPTERA) OF LEQINAT LAKES AND ADJACENT STREAMS IN BJESHKËT E NEMUNA (KOSOVO) Halil Ibrahimi1, Linda Grapci-Kotori1,*, Astrit Bilalli2, Albulena Qamili1 & Robert Schabetsberger3 1Department of Biology, Faculty of Mathematical and Natural Sciences, University of Prishtina, “Mother Theresa” p.n., 10 000 Prishtinë, Kosovo 2Faculty of Agribusiness, University of Peja “Haxhi Zeka”, UÇK street p.n., 30 000 Pejë, Kosovo 3Department of Cell Biology, Faculty of Natural Sciences, University of Salzburg, Hellbrunnerstraße 34, Raum Nr. E-2.050, Salzburg, Austria Ibrahimi, H., Grapci-Kotori, L., Bilalli, A., Qamili, A. & Schabetsberger, R.: Contribution to the knowledge of the caddisfly fauna (Insecta: Trichoptera) of Leqinat lakes and adjacent streams in Bjeshkët e Nemuna (Kosovo). Nat. Croat. Vol. 28, No. 1., 35-44, Zagreb, 2019. Adult caddisflies were collected with entomological nets and ultraviolet light traps during August and September 2018 in Leqinat Lake, Drelaj Lake and five adjacent streams in Bjeshkët e Nemuna in Kosovo. Within the current study we found three first records for the caddisfly fauna of Kosovo: Limnephilus flavospinosus, Limnephilus flavicornis and Oligotricha striata. The genus Oligotricha is reported for the first time from Kosovo. We also found few rare species which have been reported only from few localities in the Balkan Peninsula such as: Plectrocnemia mojkovacensis, Rhyacophila balcanica and Drusus tenellus. -

Vernal Pool Vernal Pool, Page 1 Michael A
Vernal Pool Vernal Pool, Page 1 Michael A. Kost Michael Overview: Vernal pools are small, isolated Introduction and Definitions: Temporary water wetlands that occur in forested settings throughout pools can occur throughout the world wherever the Michigan. Vernal pools experience cyclic periods ground or ice surface is concave and liquid water of water inundation and drying, typically filling gains temporarily exceed losses. The term “vernal with water in the spring or fall and drying during pool” has been widely applied to temporary pools the summer or in drought years. Substrates that normally reach maximum water levels in often consist of mineral soils underlain by an spring (Keeley and Zedler 1998, Colburn 2004). impermeable layer such as clay, and may be In northeastern North America, vernal pool and covered by a layer of interwoven fibrous roots similar interchangeable terms have focused even and dead leaves. Though relatively small, and more narrowly upon pools that are relatively small, sometimes overlooked, vernal pools provide critical are regularly but temporarily flooded, and are habitat for many plants and animals, including rare within wooded settings (Colburn 2004, Calhoun species and species with specialized adaptations for and deMaynadier 2008, Wisconsin DNR 2008, coping with temporary and variable hydroperiods. Ohio Vernal Pool Partnership 2009, Vermont Vernal pools are also referred to as vernal ponds, Fish & Wildlife Department 2004, Tesauro 2009, ephemeral ponds, ephemeral pools, temporary New York Natural Heritage Program 2009, pools, and seasonal wetlands. Commonwealth of Massachusetts Division of Michigan Natural Features Inventory P.O. Box 30444 - Lansing, MI 48909-7944 Phone: 517-373-1552 Vernal Pool, Page 2 Fisheries and Wildlife 2009, Maine Department of community types (see Kost et al. -

Biodiversity of Minnesota Caddisflies (Insecta: Trichoptera)
Conservation Biology Research Grants Program Division of Ecological Services Minnesota Department of Natural Resources BIODIVERSITY OF MINNESOTA CADDISFLIES (INSECTA: TRICHOPTERA) A THESIS SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY DAVID CHARLES HOUGHTON IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Ralph W. Holzenthal, Advisor August 2002 1 © David Charles Houghton 2002 2 ACKNOWLEDGEMENTS As is often the case, the research that appears here under my name only could not have possibly been accomplished without the assistance of numerous individuals. First and foremost, I sincerely appreciate the assistance of my graduate advisor, Dr. Ralph. W. Holzenthal. His enthusiasm, guidance, and support of this project made it a reality. I also extend my gratitude to my graduate committee, Drs. Leonard C. Ferrington, Jr., Roger D. Moon, and Bruce Vondracek, for their helpful ideas and advice. I appreciate the efforts of all who have collected Minnesota caddisflies and accessioned them into the University of Minnesota Insect Museum, particularly Roger J. Blahnik, Donald G. Denning, David A. Etnier, Ralph W. Holzenthal, Jolanda Huisman, David B. MacLean, Margot P. Monson, and Phil A. Nasby. I also thank David A. Etnier (University of Tennessee), Colin Favret (Illinois Natural History Survey), and Oliver S. Flint, Jr. (National Museum of Natural History) for making caddisfly collections available for my examination. The laboratory assistance of the following individuals-my undergraduate "army"-was critical to the processing of the approximately one half million caddisfly specimens examined during this study and I extend my thanks: Geoffery D. Archibald, Anne M.