From Quantum to Molecule Contents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Particle-On-A-Ring” Suppose a Diatomic Molecule Rotates in Such a Way That the Vibration of the Bond Is Unaffected by the Rotation
University of Washington Department of Chemistry Chemistry 453 Winter Quarter 2015 Lecture 17 2/25/15 Recommended Reading Atkins & DePaula 9.5 A. Background in Rotational Mechanics Two masses m1 and m2 connected by a weightless rigid “bond” of length r rotates with angular velocity rv where v is the linear velocity. As with vibrations we can render this rotation motion in simpler form by fixing one end of the bond to the origin terminating the other end with reduced mass , and allowing the reduced mass to rotate freely For two masses m1 and m2 separated by r the center of mass (CoM) is defined by the condition mr11 mr 2 2 (17.1) where m2,1 rr1,2 mm12 (17.2) Figure 17.1: Location of center of mass (CoM0 for two masses separated by a distance r. The moment of inertia I plays the role in the rotational energy that is played by mass in translational energy. The moment of inertia is 22 I mr11 mr 2 2 (17.3) Putting 17.3 into 17.4 we obtain I r 2 (17.4) mm where 12 . mm12 We can express the rotational kinetic energy K in terms of the angular momentum. The angular momentum is defined in terms of the cross product between the position vector r and the linear momentum vector p: Lrp (17.5) prprvrsin where the angle between p and r is ninety degrees for circular motion. As a cross product, the angular momentum vector L is perpendicular to the plane defined by the r and p vectors as shown in Figure 17.2 We can use equation 17.6 to obtain an expression for the kinetic energy in terms of the angular momentum L: Figure 17.2: The angular momentum L is a cross product of the position r vector and the linear momentum p=mv vector. -

Aromaticity As a Guiding Concept for Spectroscopic Features and Nonlinear Optical Properties of Porphyrinoids
molecules Article Aromaticity as a Guiding Concept for Spectroscopic Features and Nonlinear Optical Properties of Porphyrinoids Tatiana Woller 1, Paul Geerlings 1, Frank De Proft 1, Benoît Champagne 2 ID and Mercedes Alonso 1,* ID 1 Eenheid Algemene Chemie (ALGC), Vrije Universiteit Brussel (VUB), Pleinlaan 2, 1050 Brussels, Belgium; [email protected] (T.W.); [email protected] (P.G.); [email protected] (F.D.P.) 2 Laboratoire de Chimie Théorique, Unité de Chimie Physique Théorique et Structurale, University of Namur, Rue de Bruxelles 61, B-5000 Namur, Belgium; [email protected] * Correspondence: [email protected] Academic Editors: Luis R. Domingo and Miquel Solà Received: 5 March 2018; Accepted: 24 May 2018; Published: 1 June 2018 Abstract: With their versatile molecular topology and aromaticity, porphyrinoid systems combine remarkable chemistry with interesting photophysical properties and nonlinear optical properties. Hence, the field of application of porphyrinoids is very broad ranging from near-infrared dyes to opto-electronic materials. From previous experimental studies, aromaticity emerges as an important concept in determining the photophysical properties and two-photon absorption cross sections of porphyrinoids. Despite a considerable number of studies on porphyrinoids, few investigate the relationship between aromaticity, UV/vis absorption spectra and nonlinear properties. To assess such structure-property relationships, we performed a computational study focusing on a series of Hückel porphyrinoids to: (i) assess their (anti)aromatic character; (ii) determine the fingerprints of aromaticity on the UV/vis spectra; (iii) evaluate the role of aromaticity on the NLO properties. Using an extensive set of aromaticity descriptors based on energetic, magnetic, structural, reactivity and electronic criteria, the aromaticity of [4n+2] π-electron porphyrinoids was evidenced as was the antiaromaticity for [4n] π-electron systems. -
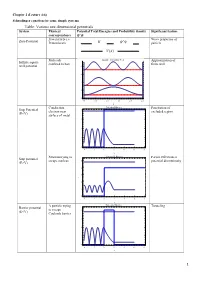
Table: Various One Dimensional Potentials System Physical Potential Total Energies and Probability Density Significant Feature Correspondence Free Particle I.E
Chapter 2 (Lecture 4-6) Schrodinger equation for some simple systems Table: Various one dimensional potentials System Physical Potential Total Energies and Probability density Significant feature correspondence Free particle i.e. Wave properties of Zero Potential Proton beam particle Molecule Infinite Potential Well Approximation of Infinite square confined to box finite well well potential 8 6 4 2 0 0.0 0.5 1.0 1.5 2.0 2.5 x Conduction Potential Barrier Penetration of Step Potential 4 electron near excluded region (E<V) surface of metal 3 2 1 0 4 2 0 2 4 6 8 x Neutron trying to Potential Barrier Partial reflection at Step potential escape nucleus 5 potential discontinuity (E>V) 4 3 2 1 0 4 2 0 2 4 6 8 x Α particle trying Potential Barrier Tunneling Barrier potential to escape 4 (E<V) Coulomb barrier 3 2 1 0 4 2 0 2 4 6 8 x 1 Electron Potential Barrier No reflection at Barrier potential 5 scattering from certain energies (E>V) negatively 4 ionized atom 3 2 1 0 4 2 0 2 4 6 8 x Neutron bound in Finite Potential Well Energy quantization Finite square well the nucleus potential 4 3 2 1 0 2 0 2 4 6 x Aromatic Degenerate quantum Particle in a ring compounds states contains atomic rings. Model the Quantization of energy Particle in a nucleus with a and degeneracy of spherical well potential which is or states zero inside the V=0 nuclear radius and infinite outside that radius. -

Course of Study
Introduction to Quantum Chemistry and Spectroscopy CY41009; Autumn Semester 2016-2017 Course of Study 1. Birth of Quantum Mechanics Failures of Classical Mechanics in Black Body Radition, Photoelectric Effects, Heat capacity of Solids, and Atomic Spectra; Quantum Mechanics - the Saviour; Birth of Quantum Chemistry. 2. Principles and Postulates of Quantum Mechanics State Functions; Operators; Eigenfunctions; Expectation Values; Time Evolution of Expectation Values; Ehrenfest Theorem; Hermitian Property; Schmidt Orthogonalization; Dirac Notation; Dirac Delta Function; Commutation of Operators; Heisenberg's Uncertainty Principle; Parity Operator. 3. Exactly Solvable Models • Translational Motions Particle in a 1D box; Bohr's Correspondence Principle; Free Particle; Particle in a 3D box; Degeneracies; Particle in a Rectangular Well; Tunneling Through Barrier; Scanning Tunneling Microscopy. • Vibrational Motions Harmonic Oscillators; Creation-Annihilation Operators; Hermite Polynomials; Vibrational Spectroscopy • Angular Motions Angular Momentum Operators; Ladder Operators; Spherical Harmonics. • Rotational Motions Particle in a ring; Particle on a sphere; Rigid Rotor; • Hydrogen Atom Solution of H-atom; Bound-state H-atom Wave Functions; Radial Distribution Functions; H-like Orbitals; Zeeman Effect; H-atom with Electron Spin; Spin-Orbit Interaction; Atomic Spectra with Spin-Orbit Interac- tion in Magnetic Field. 4. Approximate Methods Variational Theorem; He atom with Variational Method; Perturbation Theory; 1st and 2nd Order Perturbation Correction -

Quantum Mechanics Recap
Chapter 2 Quantum Mechanics in a Nutshell 2.1 Introduction 2.2 Photons Time: end of the 19th century. Maxwell’s equations have established Faraday’s hunch that light is an electromagnetic wave. However, by early 20th century, experimental evidence mounted pointing towards the fact that light is carried by ‘particles’ that pack a definite momentum and energy. Here is the crux of the problem: consider the double-slit experiment. Monochromatic light of wavelength λ passing through two slits separated by a distance d λ forms a di↵raction pattern on a photographic plate. If one ⇠ tunes down the intensity of light in a double-slit experiment, one does not get a ‘dimmer’ interference pattern, but discrete strikes on the photographic plate and illumination at specific points. That means light is composed of ‘particles’ whose energy and momentum are concentrated in one point which leads to discrete hits. But their wavelength extends over space, which leads to di↵raction patterns. Planck postulated that light is composed of discrete lumps of momemtum p = ~k and energy E = ~!. Here k =(2⇡/λ)ˆn , ˆn the direction of propagation, ~ is Planck’s constant, and ! = c k with c the speed of light. Planck’s hypothesis explained spectral | | features of the blackbody radiation. It was used by Einstein to explain the photoelectric e↵ect. Einstein was developing the theory of relativity around the same time. In this theory, the momentum of a particle of mass m and velocity v is p = mv/ 1 (v/c)2, − where c is the speed of light. Thus if a particle has m = 0, the only way itp can pack a momentum is if its velocity is v = c. -

Quantum Mechanics for Several Dimensions • Solutions for Set #10 Are Posted
Quantum Mechanics for Several Dimensions • Solutions for set #10 are posted. • Still plan a Tuesday 1-3pm session. • Some Material Covered today is not in the book- particle in ring – 2D Square Well in Chapter 8 – Coulomb Potential • Homework Set #11 is available per our vote last Friday – it is due Dec. 2. • Simple Harmonic Oscillator • Particle in a Ring -- idea of energy degeneracy • Two Dimensional Square Well – again energy degeneracy • Multiparticles in 3D http://www.colorado.edu/physics/phys2170/ Physics 2170 – Fall 2013 1 The wave functions of the first three states are Where ω = (k/m)–½ is the classical angular frequency, and n is the quantum number http://www.colorado.edu/physics/phys2170/ Physics 2170 – Fall 2013 2 http://www.colorado.edu/physics/phys2170/ Physics 2170 – Fall 2013 3 Clicker question 1 Set frequency to AD What happens to the node spacing away from x=0 for higher energy values in the harmonic oscillator potential? Think about the deBroglie wavelength. A. The node spacing doesn’t change. B. The node spacing decreases. C. The node spacing increases. http://www.colorado.edu/physics/phys2170/ Physics 2170 – Fall 2013 4 Clicker question 1 Set frequency to AD What happens to the node spacing away from x=0 for higher energy values in the harmonic oscillator potential? Think about the deBroglie wavelength. A. The node spacing doesn’t change. B. The node spacing decreases. C. The node spacing increases. http://www.colorado.edu/physics/phys2170/ Physics 2170 – Fall 2013 5 Clicker question 2 Set frequency to AD What happens to the probability density away from the equilibrium position at x=0 in the harmonic oscillator potential? A. -

Schrödinger Equation in Spherical Coordinate System and Angular
Chapter 4 (Lecture 9-10) Schrödinger Equation in Spherical coordinate system and Angular Momentum Operator In this section we will construct 3D Schrödinger equation and we give some simple examples. In this course we will consider problems where the partial differential equations are separable. Cartesian coordinate In previous chapters we have solved one dimensional problems, by using the time dependent Schrödinger equation ( ) Time part of the equation can be separated by substituting and we obtain time independent equation ( ) The equation can be extended in three dimensions (3D) by introducing 3D kinetic energy operator and potential: ( ) ( ) Where the operator ( ) Is square of gradient operator. It is obvious that 3D momentum operator can be written as ( ) Angular Momentum In quantum mechanics, the angular momentum operator is an operator analogous to classical angular momentum. ⃗ Where is position vector and is the momentum vector. The angular momentum operator plays a central role in the theory of atomic physics and other quantum problems involving rotational symmetry. In quantum mechanics angular momentum is quantized. This is because at the scale of quantum mechanics, the matter analyzed is best described by a wave equation or probability amplitude, rather than as a collection of fixed points or as a rigid body. Quantum angular momentum (cartesian) As it is known, observables in quantum physics are represented by operators. In quantum mechanics we get linear Hermitian angular momentum operators from the classical expressions using the postulates ⃗ ⃗⃗ When using Cartesian coordinates, it is customary to refer to the three spatial components of the angular momentum operator as: ( ) ( ) ( ) Square of the total angular momentum is defined as the square of the components: Commutation relation Different components of the angular momentum do not commute with another while all of the components commute with square of the total angular momentum. -
![Arxiv:2008.08022V2 [Quant-Ph] 24 Feb 2021 Time) flow “To the Left”; This Is Clearly Impossible in the Line Classical World](https://docslib.b-cdn.net/cover/1191/arxiv-2008-08022v2-quant-ph-24-feb-2021-time-ow-to-the-left-this-is-clearly-impossible-in-the-line-classical-world-2831191.webp)
Arxiv:2008.08022V2 [Quant-Ph] 24 Feb 2021 Time) flow “To the Left”; This Is Clearly Impossible in the Line Classical World
Quantum backflow in a ring Arseni Goussev School of Mathematics and Physics, University of Portsmouth, Portsmouth PO1 3HF, United Kingdom (Dated: February 25, 2021) Free motion of a quantum particle with the wave function entirely comprised of plane waves with non-negative momenta may be accompanied by negative probability current, an effect called quantum backflow. The effect is weak and fragile, and has not yet been observed experimentally. Here we show that quantum backflow becomes significantly more pronounced and more amenable to experimental observation if, instead of letting the particle move along a straight line, one forces it to move in a circular ring. I. INTRODUCTION is the so-called Bracken-Melloy bound. Finding the exact value of cline remains an open challenge. It is interesting The probability density of a quantum particle may flow to note that cline is independent of the time window T , in the direction opposite to that of the particle's momen- the particle's mass µ, or Planck's constant ~. tum [1, 2], an effect called quantum backflow (QB) [3] [4]. Many questions related to QB have been addressed The effect is inconceivable from the viewpoint of classi- in the literature. These include QB against a constant cal physics, and in this respect can be paralleled with force [7], the pertinence of QB to the arrival-time prob- other genuinely quantum phenomena such as tunneling lem [8{11], position dependence of the backflow cur- or Schr¨odinger'scat states. Unlike the latter, however, rent [5, 12, 13], probability backflow in relativistic quan- QB is relatively unexplored and yet to be observed ex- tum systems [14{16], QB in escape problems [17, 18], and perimentally. -

Quantum Corrals: Electrons in a Ring
Quantum Corrals: Electrons in a Ring Frank Rioux Chemistry Department CSB|SJU "When electrons are confined to length scales approaching the de Broglie wavelength, their behavior is dominated by quantum mechanical effects. Here we report the construction and characterization of structures for confining electrons to this length scale. The walls of these "quantum corrals" are built from Fe adatoms which are individually positioned on the Cu (111) surface by means of a scanning tunneling microscope (STM). These adatom structures confine surface state electrons laterally because of the strong scattering that occurs between surface state electrons and the Fe adatoms. The surface state electrons are confined in the direction perpendicular to the surface because of intrinsic energetic barriers that exist in that direction." This is the first paragraph of "Confinement of Electrons to Quantum Corrals on a Metal Surface," published by M. F. Crommie, C. P. Lutz, and D. M. Eigler in the October 8, 1993 issue of Science Magazine. They report the corralling of the surface electrons of Cu in a ring of radius 135 a0 created by 48 Fe adatoms. The quantum mechanics for this form of electron confinement is well-known. Schrödinger's equation for a particle in a ring and its solution (in atomic units) are given below. 2 2 1 d 1 d L Ψ()r Ψ()r Ψ()r = EΨ()r 2μ 2 2r μ dr 2 dr 2μr 2 Z nL iLθ E = Ψ ()r = JL Z R e unnormalized nL 2 nL nL 2μR th JL is the L order Bessel function, L is the angular momentum quantum number, n is the principle th quantum number, Zn,L is the n root of JL, is the effective mass of the electron, and R is the corral (ring) radius. -

Physical Chemistry II
CH354L (Unique 53470) Physical Chemistry II Time and place: Spring 2011, WAG 420, TTH 9:30-11:00AM Instructor: [email protected] ACES 4.422 232-4515 Office Hours: T 8:00-9:00AM Assistant: Michele di Pierro [email protected] Office hours (cubicle) M 4:00-6:00PM Textbook: Physical Chemistry: A Molecular Approach, by DA McQuarrie and JD Simon + lecture slides Website: Homework, lecture notes, solutions to homework and quizzes, will be posted on blackboard. Homework: Assignments will be posted on blackboard on Thursday every week. Hard copy of a solution should be handed in class on the following Thursday. You must write your own solution. Every homework assignment will include a bonus question that will be graded separately and can add up to 5 percent to the overall grade. Quizzes: Every Thursday in the last 15 minutes of the lecture there will be a quiz on material covered in the last week. Examinations: There will be three midterms that will cover non-overlapping material. The best two midterms will be used for the grades. There will also be a final examination that will cover the entire course. Grades: The grade of the course will be computed with the formula below 0.2Homeowrk + 0.05Bonus + 0.2quizzes + 0.3Midterms + 0.3Final A 85 100 B 70 85 C 55- 70 D 40- 55 F 40 +/- adjustments to grades will be given based on 15 point scale (e.g. 70-75 is B- 75-80 a B, and 80-85 a B+). The university does not allow for an A+ grade. -

Quantum Chemistry
M. Sc. IV Semester 2019-20 Paper 4305- Advanced Quantum Mechanics PARTICLE-IN-A-BOX 1. (a) Show that Aexp(ikx) Bexp(ikx) is a solution to the particle in a one- dimensional box problem. Evaluate the constants A and B. 2 Is an eigenfunction of pˆ x ? Of pˆ x ? What happens when pˆ x operates on either half of ψ? What does a solution in the form of ψ2 imply about the measurement of px? L L (b) Re-evaluate A and B placing the origin at the middle of the box, i.e. x . 2 2 Normalize the wave functions. Why are the wave functions different in this case? Would you expect any property: energies, probability density plots, expectation values of momentum, position to be different for this case? Which of these properties is different in this system? Would this affect x ? nx 2. (a) (i) Show that the function N sin satisfies the Schrödinger equation L for a particle in a one-dimensional box with a potential function V(x) equal to zero for 0 ≤ x ≤ L and infinity elsewhere. (ii) What is the eigenvalue? (iii) Describe what is meant by the Born interpretation, Pn (x) n *(x) n (x) ? (iv) What values may the quantum number n take? Sketch the first three wavefunctions and their probability density functions. (v) Determine the average position and the average momentum of the particle for an arbitrary quantum state of the particle. (vi) Show that the function is not an eigenfunction of the momentum operator d pˆ i but it is so of pˆ 2 . -

Quantum Diffusion on a Cyclic One Dimensional Lattice
Quantum diffusion on a cyclic one dimensional lattice A. C. de la Torre, H. O. M´artin, D. Goyeneche Departamento de F´ısica, Universidad Nacional de Mar del Plata Funes 3350, 7600 Mar del Plata, Argentina [email protected] (December 1, 2018) Abstract The quantum diffusion of a particle in an initially localized state on a cyclic lattice with N sites is studied. Diffusion and reconstruction time are calcu- lated. Strong differences are found for even or odd number of sites and the limit N is studied. The predictions of the model could be tested with → ∞ micro - and nanotechnology devices. I. INTRODUCTION The problem of a classical particle performing a random walk in various geometrical spaces1 and the quantum random walk2 have been thoroughly studied and compared. The classical and quantum cases have striking differences. One of these differences is that, whereas the classical spread increases with time as √T , in the quantum case we have a stronger linear time dependence of the width of the distribution. In the quantum mechanical arXiv:quant-ph/0304065v1 9 Apr 2003 case, we can identify two different causes for the spreading of the probability distribution describing the position of a particle. There is a spreading of the distribution caused by the random walk itself, also present in the classical case, and superposed to it, there is the quantum mechanical spreading of the probability distribution due to the time evolution of a particle in a localized state. This second type of spreading is the main interest of this contribution. For this study, we will consider a quantum mechanical particle initially localized in one site of a one dimensional cyclic lattice with N points.