General Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
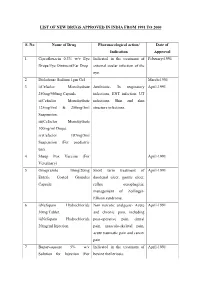
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

WHO Drug Information Vol. 12, No. 3, 1998
WHO DRUG INFORMATION VOLUME 12 NUMBER 3 • 1998 RECOMMENDED INN LIST 40 INTERNATIONAL NONPROPRIETARY NAMES FOR PHARMACEUTICAL SUBSTANCES WORLD HEALTH ORGANIZATION • GENEVA Volume 12, Number 3, 1998 World Health Organization, Geneva WHO Drug Information Contents Seratrodast and hepatic dysfunction 146 Meloxicam safety similar to other NSAIDs 147 Proxibarbal withdrawn from the market 147 General Policy Issues Cholestin an unapproved drug 147 Vigabatrin and visual defects 147 Starting materials for pharmaceutical products: safety concerns 129 Glycerol contaminated with diethylene glycol 129 ATC/DDD Classification (final) 148 Pharmaceutical excipients: certificates of analysis and vendor qualification 130 ATC/DDD Classification Quality assurance and supply of starting (temporary) 150 materials 132 Implementation of vendor certification 134 Control and safe trade in starting materials Essential Drugs for pharmaceuticals: recommendations 134 WHO Model Formulary: Immunosuppressives, antineoplastics and drugs used in palliative care Reports on Individual Drugs Immunosuppresive drugs 153 Tamoxifen in the prevention and treatment Azathioprine 153 of breast cancer 136 Ciclosporin 154 Selective serotonin re-uptake inhibitors and Cytotoxic drugs 154 withdrawal reactions 136 Asparaginase 157 Triclabendazole and fascioliasis 138 Bleomycin 157 Calcium folinate 157 Chlormethine 158 Current Topics Cisplatin 158 Reverse transcriptase activity in vaccines 140 Cyclophosphamide 158 Consumer protection and herbal remedies 141 Cytarabine 159 Indiscriminate antibiotic -

Drug Class Review Beta Adrenergic Blockers
Drug Class Review Beta Adrenergic Blockers Final Report Update 4 July 2009 Update 3: September 2007 Update 2: May 2005 Update 1: September 2004 Original Report: September 2003 The literature on this topic is scanned periodically. The purpose of this report is to make available information regarding the comparative effectiveness and safety profiles of different drugs within pharmaceutical classes. Reports are not usage guidelines, nor should they be read as an endorsement of, or recommendation for, any particular drug, use, or approach. Oregon Health & Science University does not recommend or endorse any guideline or recommendation developed by users of these reports. Mark Helfand, MD, MPH Kim Peterson, MS Vivian Christensen, PhD Tracy Dana, MLS Sujata Thakurta, MPA:HA Drug Effectiveness Review Project Marian McDonagh, PharmD, Principal Investigator Oregon Evidence-based Practice Center Mark Helfand, MD, MPH, Director Oregon Health & Science University Copyright © 2009 by Oregon Health & Science University Portland, Oregon 97239. All rights reserved. Final Report Update 4 Drug Effectiveness Review Project TABLE OF CONTENTS INTRODUCTION .......................................................................................................................... 6 Purpose and Limitations of Evidence Reports........................................................................................ 8 Scope and Key Questions .................................................................................................................... 10 METHODS................................................................................................................................. -

IIIHIIIUSOO5304377A Unitedo States Patent 19 (11) Patent Number: 5,304,377 Yamada Et Al
IIIHIIIUSOO5304377A UnitedO States Patent 19 (11) Patent Number: 5,304,377 Yamada et al. (45) Date of Patent: Apr. 19, 1994 54 PROLONGED RELEASE PREPARATION 4,954,298 9/1990 Yamamoto et al. ................. 264/4.6 AND POLYMERS THEREOF 4,962,091 10/1990 Eppstein et al. ........................ 514/2 75) Inventors: Minoru Yamada; Seiko Ishiguro, both 5,061,492 10/1991 Okada et al. ........................ 424/423 Otokuni, all of Japan 0052510 11/1981 European Pat. Off. 73) Assignee: Sherical4. industries, Ltd., O256726O190833 7/19871/1986 European Pat. Off. a, Japan 0263490 10/1987 European Pat. Off. (21) Appl. No.: 986,299 0281482 3/1988 European Pat. Off. 035.0246 7/1989 European Pat. Off. 22 Filed: Dec. 7, 1992 2-212436 8/1990 Japan. Related U.S. Application Data OTHER PUBLICATIONS 63) continuation of ser. No. 777,170, oct. 16, 1991, aban- Chemical Abstracts, vol. 114/No. 6 (Feb. 11, 1991); doned. Columbus, Ohio; Abstract No. 49615R. 30 Foreign Application Priority Data Primary Ent, S. Sir Mueller & Oct. 16, 1990 (JP) Japan .................................. 2-278037 story Agent, or Firm-Wegner, Cantor, Mueller Aug. 28, 1991 JP Japan .................................. 3.217045 51 Int, Cl. ......................... A61K 9/22; A61K 9/52; ' ABSTRACT A61K 31/74 A polymer for a prolonged release preparation which 52 U.S. Cl. ................................. 424/426; 424/78.08; comprises 424/78.37; 424/434; 424/457; 424/468; (A) a polylactic acid and 424/486; 424/497; 525/450, 514/2 (B) a copolymer of glycolic acid and a hydroxycar 58) Field of Search .................. 424/426, 78.08, 78.37, boxylic acid of general formula 424/434, 457, 468, 486, 497; 525/450; 514/2 (56) References Cited R U.S. -

Spectrofluorometric Determination of Some Β-Blockers in Tablets And
ORIGINAL ARTICLES Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia Spectrofluorometric determination of some b-blockers in tablets and human plasma using 9,10-dimethoxyanthracene-2-sodium sulfonate H. Abdine, M. A. Sultan, M. M. Hefnawy, F. Belal Received April 16, 2004, accepted June 2, 2004 Prof. Dr. F. Belal, Department of Pharmaceutical Chemistry, College of Pharmacy, P.O.Box 2457, King Saud University, Riyadh 11451, Saudi Arabia [email protected] Pharmazie 60: 265–268 (2005) A simple and sensitive spectrofluorometric method was developed for the quantitative determination of some b-blockers, namely arotinolol, atenolol and labetalol as hydrochloride salts. The method is based on the reaction of these drugs as n-electron donors with the fluorogenic reagent 9,10-dimethoxy-2- anthracene sulfonate (DMAS) as p-acceptor in acidic medium. The obtained ion-pairs were extracted into chloroform and measured spectrofluorometrically at 452 nm after excitation at 385 nm. The fluor- escence intensity-concentration plots are rectilinear over the ranges of 0.5–5 mg Á ml1, 1.0–11.0 mg Á ml1 and 0.6–6.4 mg Á ml1 for labetalol, atenolol and arotinolol, respectively. The different parameters affect- ing the reaction pathway were thoroughly studied and optimized. No interference was observed from the common pharmaceutical excipients. The proposed method was successfully applied to the analy- sis of tablets and the results were statistically compared with those obtained by reference methods. The method was further extended to the in vitro determination of the drugs in spiked human plasma, the% recoveries (n ¼ 3) ranged from 96.98 Æ 1.55 to 98.28 Æ 2.19. -

A Comparative Study of DA-9601 and Misoprostol for Prevention of NSAID-Associated Gastroduodenal Injury in Patients Undergoing Chronic NSAID Treatment
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector Arch. Pharm. Res. (2014) 37:1308–1316 DOI 10.1007/s12272-014-0408-3 RESEARCH ARTICLE A comparative study of DA-9601 and misoprostol for prevention of NSAID-associated gastroduodenal injury in patients undergoing chronic NSAID treatment Oh Young Lee • Dae-Hwan Kang • Dong Ho Lee • Il-Kwun Chung • Jae-Young Jang • Jin-Il Kim • Jin-Woong Cho • Jong-Sun Rew • Kang-Moon Lee • Kyoung Oh Kim • Myung-Gyu Choi • Sang-Woo Lee • Soo-Teik Lee • Tae-Oh Kim • Yong-Woon Shin • Sang-Yong Seol Received: 25 April 2014 / Accepted: 2 May 2014 / Published online: 30 May 2014 Ó The Author(s) 2014. This article is published with open access at Springerlink.com Abstract Misoprostol is reported to prevent non-steroidal The primary endpoint was the gastric protection rate, and anti-inflammatory drug (NSAID)-associated gastroduode- secondary endpoints were the duodenal protection rate and nal complications. There is, however, limited information ulcer incidence rate. Endpoints were assessed by endos- regarding the efficacy of DA-9601 in this context. We copy after the 4-week treatment period. Drug-related performed a comparative study on the relative efficacy of adverse effects, including gastrointestinal (GI) symptoms, DA-9601 and misoprostol for prevention of NSAID-asso- were also compared. At week 4, the gastric protection rates ciated complications. In this multicenter, double-blinded, with DA-9601 and misoprostol were 81.4 % (192/236) and active-controlled, stratified randomized, parallel group, 89.3 % (216/242), respectively. -

Dorset Medicines Advisory Group
DORSET CARDIOLOGY WORKING GROUP GUIDELINE FOR CALCIUM CHANNEL BLOCKERS IN HYPERTENSION SUMMARY The pan-Dorset cardiology working group continues to recommend the use of amlodipine (a third generation dihydropyridine calcium-channel blocker) as first choice calcium channel blocker on the pan-Dorset formulary for hypertension. Lercanidipine is second choice, lacidipine third choice and felodipine is fourth choice. This is due to preferable side effect profiles in terms of ankle oedema and relative costs of the preparations. Note: where angina is the primary indication or is a co-morbidity prescribers must check against the specific product characteristics (SPC) for an individual drug to confirm this is a licensed indication. N.B. Lacidipine and lercandipine are only licensed for use in hypertension. Chapter 02.06.02 CCBs section of the Formulary has undergone an evidence-based review. A comprehensive literature search was carried out on NHS Evidence, Medline, EMBASE, Cochrane Database, and UK Duets. This was for recent reviews or meta-analyses on calcium channel blockers from 2009 onwards (comparative efficacy and side effects) and randomised controlled trials (RCTs). REVIEW BACKGROUND Very little good quality evidence exists. No reviews, meta-analyses or RCTs were found covering all calcium channel blockers currently on the formulary. Another limitation was difficulty obtaining full text original papers for some of the references therefore having to use those from more obscure journals instead. Some discrepancies exist between classification of generations of dihydropyridine CCBs, depending upon the year of publication of the reference/authors’ interpretation. Dihydropyridine (DHP) CCBs tend to be more potent vasodilators than non-dihydropyridine (non-DHP) CCBs (diltiazem, verapamil), but the latter have greater inotropic effects. -

A Meta-Analysis of the Efficacy and Safety of Arotinolol in the Treatment of Chinese Patients with Essential Hypertension
African Journal of Pharmacy and Pharmacology Vol. 6(1), pp. 36-42, 8 January, 2011 Available online at http://www.academicjournals.org/AJPP DOI: 10.5897/AJPP11.452 ISSN 1996-0816 ©2011 Academic Journals Full Length Research Paper A meta-analysis of the efficacy and safety of arotinolol in the treatment of Chinese patients with essential hypertension Du Bing 1# ,,,Cui Wen-peng 2# ,,,Xu Guo-liang 1, Sun Guo-qiang 1, Wu Guo-dong 1, Qu Rui 1, Lin Shu-mei 1, Liu Yun-yang 1, Qin Ling 1* 1Department of Cardiology, the second part of First Hospital, Jilin University, Changchun, China. 2Department of Nephrology, Second Hospital, Jilin University, Changchun, China. Accepted 8 September, 2011 Arotinolol had been used for treatment of essential hypertension. We conducted a meta-analysis to compare the efficacy and safety of arotinolol with other antihypertensive drugs in treating essential hypertension. Medical databases and review articles were screened with prespecified criteria for randomized controlled trials that reported the effects of and adverse reactions to arotinolol and other antihypertensive drugs in treating essential hypertension. Literature identified meta-analysed using RevMan4.2. Methodology quality of the selected studies was conducted using a Jadad scale. The results were that a total of 176 articles had been found of which 6 were finally included for meta- analysis. The meta-analysis compared the efficacy and safety of arotinolol with other common antihypertensive drugs, including enalapril, felodipine, imidapril, cilinidipine, metroprolol and atenolol. Results indicated that there were no evidence for differences in safety and efficacy. Homogeneity test, χ2 = 4.41, df = 7, P = 0.73 (efficacy); χ2 = 2.96, df = 4, P = 0.56 (safety); combined test, Z = 0.64 (P = 0.52), OR = 1.17, 95% confidence interval (CI) (0.72, 1.85) (efficacy); Z = 1.75 (P = 0.08), OR = 0.60, 95% CI (0.34, 1.06) (safety). -

Customs Tariff - Schedule
CUSTOMS TARIFF - SCHEDULE 99 - i Chapter 99 SPECIAL CLASSIFICATION PROVISIONS - COMMERCIAL Notes. 1. The provisions of this Chapter are not subject to the rule of specificity in General Interpretative Rule 3 (a). 2. Goods which may be classified under the provisions of Chapter 99, if also eligible for classification under the provisions of Chapter 98, shall be classified in Chapter 98. 3. Goods may be classified under a tariff item in this Chapter and be entitled to the Most-Favoured-Nation Tariff or a preferential tariff rate of customs duty under this Chapter that applies to those goods according to the tariff treatment applicable to their country of origin only after classification under a tariff item in Chapters 1 to 97 has been determined and the conditions of any Chapter 99 provision and any applicable regulations or orders in relation thereto have been met. 4. The words and expressions used in this Chapter have the same meaning as in Chapters 1 to 97. Issued January 1, 2020 99 - 1 CUSTOMS TARIFF - SCHEDULE Tariff Unit of MFN Applicable SS Description of Goods Item Meas. Tariff Preferential Tariffs 9901.00.00 Articles and materials for use in the manufacture or repair of the Free CCCT, LDCT, GPT, UST, following to be employed in commercial fishing or the commercial MT, MUST, CIAT, CT, harvesting of marine plants: CRT, IT, NT, SLT, PT, COLT, JT, PAT, HNT, Artificial bait; KRT, CEUT, UAT, CPTPT: Free Carapace measures; Cordage, fishing lines (including marlines), rope and twine, of a circumference not exceeding 38 mm; Devices for keeping nets open; Fish hooks; Fishing nets and netting; Jiggers; Line floats; Lobster traps; Lures; Marker buoys of any material excluding wood; Net floats; Scallop drag nets; Spat collectors and collector holders; Swivels. -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Genl:VE 1970 © World Health Organization 1970
Nathan B. Eddy, Hans Friebel, Klaus-Jiirgen Hahn & Hans Halbach WORLD HEALTH ORGANIZATION ORGANISATION .MONDIALE DE LA SANT~ GENl:VE 1970 © World Health Organization 1970 Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Convention. Nevertheless governmental agencies or learned and professional societies may reproduce data or excerpts or illustrations from them without requesting an authorization from the World Health Organization. For rights of reproduction or translation of WHO publications in toto, application should be made to the Division of Editorial and Reference Services, World Health Organization, Geneva, Switzerland. The World Health Organization welcomes such applications. Authors alone are responsible for views expressed in signed articles. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Director-General of the World Health Organization concerning the legal status of any country or territory or of its authorities, or concerning the delimitation of its frontiers. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. © Organisation mondiale de la Sante 1970 Les publications de l'Organisation mondiale de la Sante beneficient de la protection prevue par les dispositions du Protocole n° 2 de la Convention universelle pour la Protection du Droit d'Auteur. Les institutions gouvernementales et les societes savantes ou professionnelles peuvent, toutefois, reproduire des donnees, des extraits ou des illustrations provenant de ces publications, sans en demander l'autorisation a l'Organisation mondiale de la Sante. Pour toute reproduction ou traduction integrate, une autorisation doit etre demandee a la Division des Services d'Edition et de Documentation, Organisation mondiale de la Sante, Geneve, Suisse. -

(19) United States (12) Patent Application Publication (10) Pub
US 20050181041A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0181041 A1 Goldman (43) Pub. Date: Aug. 18, 2005 (54) METHOD OF PREPARATION OF MIXED Related US. Application Data PHASE CO-CRYSTALS WITH ACTIVE AGENTS (60) Provisional application No. 60/528,232, ?led on Dec. 9, 2003. Provisional application No. 60/559,862, ?led (75) Inventor: David Goldman, Portland, CT (US) on Apr. 6, 2004. Correspondence Address: Publication Classi?cation LEYDIG VOIT & MAYER, LTD (51) Int. Cl.7 ....................... .. A61K 31/56; A61K 38/00; TWO PRUDENTIAL PLAZA, SUITE 4900 A61K 9/64 180 NORTH STETSON AVENUE (52) US. Cl. ............................ .. 424/456; 514/179; 514/2; CHICAGO, IL 60601-6780 (US) 514/221 (73) Assignee: MedCrystalForms, LLC, Hunt Valley, (57) ABSTRACT MD This invention pertains to a method of preparing mixed phase co-crystals of active agents With one or more materials (21) Appl. No.: 11/008,034 that alloWs the modi?cation of the active agent to a neW physical/crystal form With unique properties useful for the delivery of the active agent, as Well as compositions com (22) Filed: Dec. 9, 2004 prising the mixed phase co-crystals. Patent Application Publication Aug. 18, 2005 Sheet 1 0f 8 US 2005/0181041 A1 FIG. 1a 214.70°C z.m."m.n... 206.98°C n..0ao 142 OJ/g as:20m=3: -0.8 -1.0 40 90 1:10 2110 Temperture (°C) FIG. 1b 0.01 as:22“.Km: 217 095 24221.4 39Jmum/Q -0.8 35 155 255 255 Temperture (°C) Patent Application Publication Aug.