Spectrofluorometric Determination of Some Β-Blockers in Tablets And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Drug Class Review Beta Adrenergic Blockers
Drug Class Review Beta Adrenergic Blockers Final Report Update 4 July 2009 Update 3: September 2007 Update 2: May 2005 Update 1: September 2004 Original Report: September 2003 The literature on this topic is scanned periodically. The purpose of this report is to make available information regarding the comparative effectiveness and safety profiles of different drugs within pharmaceutical classes. Reports are not usage guidelines, nor should they be read as an endorsement of, or recommendation for, any particular drug, use, or approach. Oregon Health & Science University does not recommend or endorse any guideline or recommendation developed by users of these reports. Mark Helfand, MD, MPH Kim Peterson, MS Vivian Christensen, PhD Tracy Dana, MLS Sujata Thakurta, MPA:HA Drug Effectiveness Review Project Marian McDonagh, PharmD, Principal Investigator Oregon Evidence-based Practice Center Mark Helfand, MD, MPH, Director Oregon Health & Science University Copyright © 2009 by Oregon Health & Science University Portland, Oregon 97239. All rights reserved. Final Report Update 4 Drug Effectiveness Review Project TABLE OF CONTENTS INTRODUCTION .......................................................................................................................... 6 Purpose and Limitations of Evidence Reports........................................................................................ 8 Scope and Key Questions .................................................................................................................... 10 METHODS................................................................................................................................. -

A Meta-Analysis of the Efficacy and Safety of Arotinolol in the Treatment of Chinese Patients with Essential Hypertension
African Journal of Pharmacy and Pharmacology Vol. 6(1), pp. 36-42, 8 January, 2011 Available online at http://www.academicjournals.org/AJPP DOI: 10.5897/AJPP11.452 ISSN 1996-0816 ©2011 Academic Journals Full Length Research Paper A meta-analysis of the efficacy and safety of arotinolol in the treatment of Chinese patients with essential hypertension Du Bing 1# ,,,Cui Wen-peng 2# ,,,Xu Guo-liang 1, Sun Guo-qiang 1, Wu Guo-dong 1, Qu Rui 1, Lin Shu-mei 1, Liu Yun-yang 1, Qin Ling 1* 1Department of Cardiology, the second part of First Hospital, Jilin University, Changchun, China. 2Department of Nephrology, Second Hospital, Jilin University, Changchun, China. Accepted 8 September, 2011 Arotinolol had been used for treatment of essential hypertension. We conducted a meta-analysis to compare the efficacy and safety of arotinolol with other antihypertensive drugs in treating essential hypertension. Medical databases and review articles were screened with prespecified criteria for randomized controlled trials that reported the effects of and adverse reactions to arotinolol and other antihypertensive drugs in treating essential hypertension. Literature identified meta-analysed using RevMan4.2. Methodology quality of the selected studies was conducted using a Jadad scale. The results were that a total of 176 articles had been found of which 6 were finally included for meta- analysis. The meta-analysis compared the efficacy and safety of arotinolol with other common antihypertensive drugs, including enalapril, felodipine, imidapril, cilinidipine, metroprolol and atenolol. Results indicated that there were no evidence for differences in safety and efficacy. Homogeneity test, χ2 = 4.41, df = 7, P = 0.73 (efficacy); χ2 = 2.96, df = 4, P = 0.56 (safety); combined test, Z = 0.64 (P = 0.52), OR = 1.17, 95% confidence interval (CI) (0.72, 1.85) (efficacy); Z = 1.75 (P = 0.08), OR = 0.60, 95% CI (0.34, 1.06) (safety). -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Essential Tremor: Diagnosis and Management BMJ: First Published As 10.1136/Bmj.L4485 on 5 August 2019
STATE OF THE ART REVIEW Essential tremor: diagnosis and management BMJ: first published as 10.1136/bmj.l4485 on 5 August 2019. Downloaded from Vicki Shanker ABSTRACT Essential tremor is one of the most common movement disorders in adults and can Department of Neurology, Icahn School of Medicine at Mount affect both children and adults. An updated consensus statement in 2018 redefined Sinai, New York, NY 10029, USA essential tremor as an isolated action tremor present in bilateral upper extremities Correspondence to: [email protected] for at least three years. Tremor may also be present in other locations, commonly Additional material is published the neck or the vocal cords. Patients with additional neurologic symptoms are online only. To view please visit the journal online. now categorized as “essential tremor plus.” Additional clinical features associated Cite this as: BMJ 2019;366:l4485 with the condition include but are not limited to cognitive impairment, psychiatric http://dx.doi.org/10.1136/bmj.l4485 Series explanation: State of the disorders, and hearing loss. When treatment is needed, propranolol and primidone Art Reviews are commissioned are considered first line treatments. Patients who are severely affected are often on the basis of their relevance to academics and specialists offered deep brain stimulation. Although the ventral intermediate nucleus of the in the US and internationally. For this reason they are written thalamus is the traditional surgical target, the caudal zona incerta is also being predominantly by US authors. studied as a possible superior alternative. Magnetic resonance imaging guided high intensity focused ultrasound is a newer surgical alternative that may be ideal for patients with substantial medical comorbidities. -
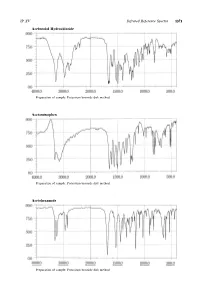
Infrared Reference Spectra a to B
JP XV Infrared Reference Spectra 1373 Acebutolol Hydrochloride Preparation of sample: Potassium bromide disk method Acetaminophen Preparation of sample: Potassium bromide disk method Acetohexamide Preparation of sample: Potassium bromide disk method 1374 Infrared Reference Spectra JP XV Acetylcholine Chloride for Injection Preparation of sample: Potassium bromide disk method Aclarubicin Hydrochloride Preparation of sample: Potassium bromide disk method Acrinol Hydrate Preparation of sample: Potassium bromide disk method JP XV Infrared Reference Spectra 1375 Afloqualone Preparation of sample: Potassium bromide disk method Alacepril Preparation of sample: Potassium bromide disk method Alprenolol Hydrochloride Preparation of sample: Potassium chloride disk method 1376 Infrared Reference Spectra JP XV Alprostadil Preparation of sample: Potassium bromide disk method Amantadine Hydrochloride Preparation of sample: Potassium chloride disk method Ambenonium Chloride Preparation of sample: Potassium chloride disk method JP XV Infrared Reference Spectra 1377 Amidotrizoic Acid Preparation of sample: Potassium bromide disk method Amikacin Sulfate Preparation of sample: Potassium bromide disk method Amoxapine Preparation of sample: Potassium bromide disk method 1378 Infrared Reference Spectra JP XV Amoxicillin Hydrate Preparation of sample: Potassium bromide disk method Anhydrous Ampicillin Preparation of sample: Potassium bromide disk method Ampicillin Hydrate Preparation of sample: Potassium bromide disk method JP XV Infrared Reference Spectra 1379 -

Acebutolol Hydrochloride Acetohexamide 1 Acetohexamide 2
JP XV Ultraviolet-visible Reference Spectra 1519 Acebutolol Hydrochloride A solution in 0.01 mol/L hydrochloric acid TS (1 in 100,000) Acetohexamide 1 A solution prepared as follows: Dissolve 0.10 g in 100 mL of methanol. To 5 mL of this solution add 20 mL of 0.5 mol/L hydrochloric acid TS and 75 mL of methanol (Sample solution 1). Acetohexamide 2 A solution prepared as follows: To 10 mL of Sample solution 1 add methanol to make 50 mL (Sample solution 2). 1520 Ultraviolet-visible Reference Spectra JP XV Aclarubicin Hydrochloride A solution in diluted methanol (4 in 5) (3 in 100,000) Acrinol Hydrate An aqueous solution (3 in 250,000) Actinomycin D A solution in methanol (3 in 100,000) JP XV Ultraviolet-visible Reference Spectra 1521 Afloqualone A solution in ethanol (99.5) (1 in 150,000) Alimemazine Tartrate An aqueous solution (1 in 100,000) Allopurinol An aqueous solution (1 in 200,000) 1522 Ultraviolet-visible Reference Spectra JP XV Alprazolam A solution in ethanol (95) (1 in 200,000) Alprenolol Hydrochloride A solution in ethanol (95) (1 in 10,000) Alprostadil A solution prepared as follows: To 10 mL of a solution in ethanol (99.5) (1 in 100,000) add 1 mL of potassium hydroxide-ethanol TS, and allow to stand for 15 minutes. JP XV Ultraviolet-visible Reference Spectra 1523 Alprostadil Alfadex A solution prepared as follows: To 10 mL of a solution in dilute ethanol (3 in 10,000) add 1 mL of potassium hydroxide-ethanol TS, and allow to stand for 15 minutes. -

Drugs for Primary Prevention of Atherosclerotic Cardiovascular Disease: an Overview of Systematic Reviews
Supplementary Online Content Karmali KN, Lloyd-Jones DM, Berendsen MA, et al. Drugs for primary prevention of atherosclerotic cardiovascular disease: an overview of systematic reviews. JAMA Cardiol. Published online April 27, 2016. doi:10.1001/jamacardio.2016.0218. eAppendix 1. Search Documentation Details eAppendix 2. Background, Methods, and Results of Systematic Review of Combination Drug Therapy to Evaluate for Potential Interaction of Effects eAppendix 3. PRISMA Flow Charts for Each Drug Class and Detailed Systematic Review Characteristics and Summary of Included Systematic Reviews and Meta-analyses eAppendix 4. List of Excluded Studies and Reasons for Exclusion This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. 1 Downloaded From: https://jamanetwork.com/ on 09/28/2021 eAppendix 1. Search Documentation Details. Database Organizing body Purpose Pros Cons Cochrane Cochrane Library in Database of all available -Curated by the Cochrane -Content is limited to Database of the United Kingdom systematic reviews and Collaboration reviews completed Systematic (UK) protocols published by by the Cochrane Reviews the Cochrane -Only systematic reviews Collaboration Collaboration and systematic review protocols Database of National Health Collection of structured -Curated by Centre for -Only provides Abstracts of Services (NHS) abstracts and Reviews and Dissemination structured abstracts Reviews of Centre for Reviews bibliographic -

Beneficial Effects of Γ-Aminobutyric Acid on Right Ventricular Pressure
Life Sciences 91 (2012) 693–698 Contents lists available at SciVerse ScienceDirect Life Sciences journal homepage: www.elsevier.com/locate/lifescie Beneficial effects of γ-aminobutyric acid on right ventricular pressure and pulmonary vascular remodeling in experimental pulmonary hypertension Rie Suzuki a, Rumi Maehara a, Shuhei Kobuchi b, Ryosuke Tanaka a, Mamoru Ohkita a, Yasuo Matsumura a,⁎ a Laboratory of Pathological and Molecular Pharmacology, Osaka University of Pharmaceutical Sciences, 4-20-1 Nasahara, Takatsuki, Osaka, Japan b Department of Pharmacy, School of Pharmacy, Hyogo University of Health Sciences, Kobe, Hyogo, Japan article info abstract Article history: Aims: It has been reported that activation of the sympathetic nervous system and increase in plasma Received 19 October 2011 norepinephrine (NE) levels are observed in patients with pulmonary hypertension (PH). γ-Aminobutyric Accepted 4 April 2012 acid (GABA) is one of the major inhibitory neurotransmitters in the central nervous system and suppresses peripheral sympathetic neurotransmission. This study investigated whether chronic treatment with GABA Keywords: prevents the development of monocrotaline (MCT)-induced PH. To elucidate the relationship between Monocrotaline the development of PH and sympathetic nerve activity, hemodynamic parameters, cardiac functions, and Pulmonary hypertension γ-Aminobutyric acid plasma NE concentrations as well as cardiac endothelin-1 (ET-1) contents of MCT-induced PH rats were Norepinephrine evaluated with or without GABA treatment. Sympathetic nervous system Main methods: Rats were injected with MCT (60 mg/kg) or saline subcutaneously and these rats were Endothelin-1 randomly divided into GABA (500 mg/kg/day for 4 weeks)- or vehicle-treated groups, respectively. Key finding: MCT-treated rats had higher right ventricular systolic pressures, right ventricle-to-left ventricle plus septum weight ratios, pulmonary arterial medial thickening, and plasma NE levels than those of saline-injected rats. -

The Advantages of Carvedilol in Coronary Artery Disease
FACTA UNIVERSITATIS Series: Medicine and Biology Vol.8, No 1, 2001, pp. 11 - 18 UC 612.17; 611.12 THE ADVANTAGES OF CARVEDILOL IN CORONARY ARTERY DISEASE Svetlana Apostolović¹, Milan Pavlović¹, Miloje Tomašević¹, Aleksandar Stojković¹, Ružica Janković¹, Slobodan Ilić² ¹Clinic for Cardiology, Clinical Center, Niš, Yugoslavia, ²Society for Liason Psychiatry, Niš, Yugoslavia E-mail: [email protected] Summary. Dilatrend (carvedilol) is a nonselective beta and alpha blocker with antioxidation and antiprolipherative effects. These effects prevent mechanical damage of the endothelium of coronary arteries and have no negative influ- ence on metabolic risk factors: lipid prophyl and insulin resistence. With beta-blockade, dilatrend (carvedilol) reduces the heart rate and blood pressure, with alpha-blockade it reduces the peripheral vascular resistence and in that way decreases the myocardial oxygen consumption and myocardial wall stress. Dilatrend (carvedilol) prolonges exercise limits (duration of exercise). Applied in the acute phase of myocardial infarction, dilatrend (carvedilol) increases the patients' survival rate, reduces new coronary events, and with antioxydation action it prevents reperfusion arrhyth- mias. With positive hymodinamic effects it improves the left ventricle function in patients with ischemic cardiomyopa- thy, and gives a chance for survival to the coronary patients who run high surgery risk. Key words: Carvedilol, coronary disease, myocardial infarction The effect of carvedilol we strive to maintain the function and the integrity of upon the coronary arteries endothelium the endothelium of coronary arteries, primarily by controling the variable risk factors. Today, by using a The central position in the onset and the determina- particular drug it is possible to influence most of the tion of the course of the coronary disease is occupied by mentioned processes that lead to the damage of the the atherosclerotic plaque of the coronary artery. -

Reporting and Learning System for Medication Errors: the Role Of
REPORTING AND LEARNING SYSTEMS FOR MEDICATION ErrORS: THE ROLE OF PHARMACOVIGILANCE CENTRES Reporting and learning systems for medication errors: the role of pharmacovigilance centres WHO Library Cataloguing-in-Publication Data Reporting and learning systems for medication errors: the role of pharmacovigilance centres. 1.Medication Errors – prevention and control. 2.Pharmacovigilance. 3.Drug Monitoring. I.World Health Organization. ISBN 978 92 4 150794 3 (NLM classification: QV 771) © World Health Organization 2014 All rights reserved. Publications of the World Health Organization are available on the WHO website (www.who.int) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publica- tions – whether for sale or for non-commercial distribution – should be addressed to WHO Press through the WHO website (www.who.int/about/licensing/copyright_form/en/index.html). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization con- cerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted and dashed lines on maps represent ap- proximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. -

Predicting Interactions of Antihypertensive Drugs and Rifampin in Tuberculosis Patients Using BDDCS Tingting Yan1,2#, Wei Liu1,3#, Ken Chen1,2, Li Yang1,3, Leslie Z
Predicting interactions of antihypertensive drugs and rifampin in tuberculosis patients using BDDCS Tingting Yan1,2#, Wei Liu1,3#, Ken Chen1,2, Li Yang1,3, Leslie Z. Benet1,4*, Suodi Zhai1,3* 1 Pharmacy Department, Peking University Third Hospital, Beijing 100191, China; 2 Department of Pharmacy Administration and Clinical Pharmacy, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China; 3 Peking University Therapeutic Drug Monitoring and Clinical Toxicology Center, Beijing 100191 China; 4 University of California San Francisco, San Francisco, 94143, United States #Tingting Yan and Wei Liu contributed equally to this work. *Corresponding author at: Peking University Third Hospital, No. 49 North Garden Rd, Haidian District, Beijing 100191, China (Suodi Zhai); University of California San Francisco, 533 Parnassus Avenue, Room U-68, San Francisco, CA 94143- 0912, USA (Leslie Z. Benet). Tel: 8610-82266686 (Suodi Zhai); (415) 476-3853 (Leslie Z. Benet) Fax: 8610-82266686 (Suodi Zhai); (415) 476-8887 (Leslie Z. Benet) E-mail: [email protected] (Suodi Zhai); [email protected] (Leslie Z. Benet) Running Head: Predicting interactions of antihypertensive drugs and rifampin in tuberculosis patients using BDDCS 1 Abstract Background Lack of blood pressure control is often seen in hypertensive patients concomitantly taking antituberculosis (TB) medications due to the complex drug-drug interactions between rifampin and antihypertensive drugs. Therefore, it is of clinical importance to understand the mechanism of interactions between rifampin and antihypertensive drugs, thereby allowing predictions and recommendations on the use of antihypertensive drugs in such co-medicated patients. Objective To predict interactions between antihypertensive drugs and rifampin under the theory of the Biopharmaceutics Drug Disposition Classification System (BDDCS), taking into consideration the role of drug transporter and metabolic enzyme interactions, and to give guidance on the selection of antihypertensive drugs for patients with TB. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.